
Expired activity
Please go to the PowerPak
homepage and select a course.
Improving Outcomes in Spinal Muscular Atrophy: What Pharmacists Need to Know
EPIDEMIOLOGY/PATHOPHYSIOLOGY
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease that affects 1 in 11,000 births.1 Its prevalence is approximately 1 to 2 in 100,000 people, affecting fewer than 9,000 patients in the United States, Europe, and Australia combined.2,3 The progression of SMA results in muscle weakness, paralysis, and premature death. Without treatment, the life expectancy with SMA could be anywhere from less than 1 month to a normal adult life expectancy, depending on disease severity.4
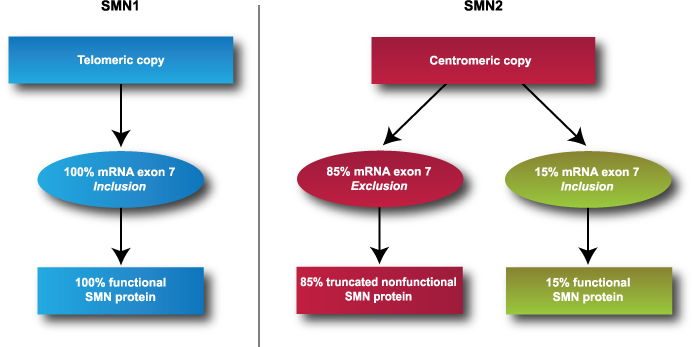
SMA is caused by a deletion or mutation in the survival motor neuron 1 (SMN1) gene found on chromosome 5. The SMN1 gene contains instructions for making the SMN protein, which is part of a protein complex needed to process pre-mRNA and create and maintain motor neuron cells.5 In addition to SMN1, humans have a nearly identical, SMN2 gene, which also produces SMN proteins. However, approximately 85% of SMN2 transcription leads to truncated and nonfunctional SMN proteins due to splicing, or skipping, of exon 7 (see Figure 1). A healthy individual typically has 2 copies of SMN1 and 1 to 2 copies of SMN2 (although SMN2 copy number can vary up to 8). The more copies of SMN2, the greater the quantity of functional SMN proteins that can be produced.
Motor neurons are specialized nerve cells that can be found in tissue types throughout the body, with the highest levels in the spinal cord.6 Motor neurons transmit signals from the brain and spinal cord to skeletal muscle to cause muscle contractions and movement. Due to the deletion or mutation in the SMN1 gene and subsequent motor neuron loss, SMA patients present with muscle weakness and atrophy that worsens with age. The most notable weakness is typically symmetrical and located in proximal limbs.7 SMA can also lead to downstream complications of other organ systems such as respiratory distress, cardiac complications, and gastrointestinal dysfunction.8
Figure 1. Genetic expression of survival motor neuron genes 1 and 2.
DIAGNOSIS/CLASSIFICATION
Currently, methods for detecting SMA include carrier testing, prenatal and newborn screening, and genetic testing. Carrier screening can detect whether a parent has only one healthy copy of SMN1, which means the other copy is faulty and could potentially be passed to a child. Carrier frequency has been reported to be 1 in 47 people in the North American population. 9
A prenatal screen is also available during pregnancy via chorionic villus sampling or amniocentesis to obtain fetal DNA, though this option is not frequently used. More commonly, infants are tested after birth as part of the newborn screen.10 SMA was recently added to the federal Recommended Uniform Screening Panel in 2018. This test is able to detect 95% of babies with SMA, but to date only 31 states have added SMA to their newborn screen (see Figure 2).11
The final method to detect SMA is through recognition of clinical signs and symptoms and confirmation via molecular genetic testing. SMA typically presents with hypotonia, progressive symmetric and proximal muscle weakness, hyporeflexia, and tongue fasciculations. Genetic testing using multiplex ligation–dependent probe amplification (MLPA) or quantitative polymerase chain reaction (qPCR) can identify exon 7 deletion on the SMN1 gene as well as SMN1 and SMN2 copy number.11
Figure 2. Prenatal screening for SMA availability in the United States12
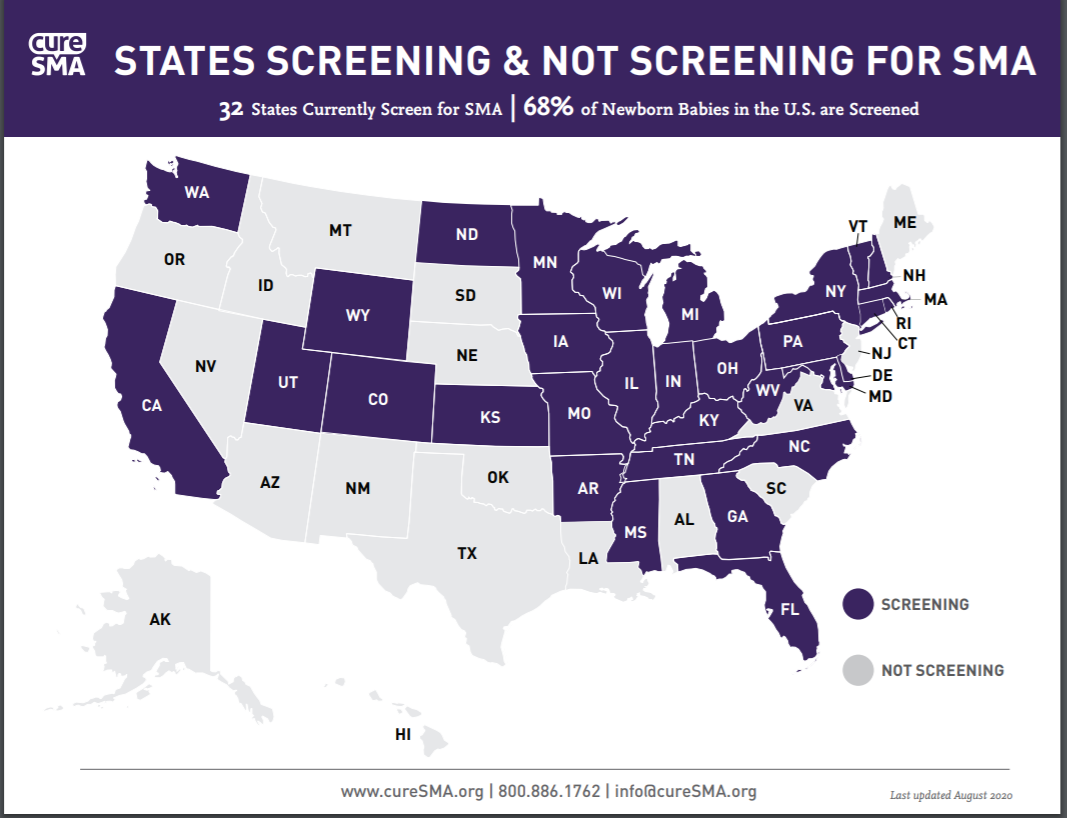
Source: Reprinted with permission of Cure SMA. All rights reserved.
SMA has historically been classified as types 0 through 4 based on age of onset, highest physical function, as well as number of SMN2 copies. Generally, an earlier age of onset and a lower number of SMN2 copies may be indicative of more severe disease (see Table 1). SMA type 0 is the most severe form with prenatal onset of symptoms and a life expectancy of less than 1 month of age. SMA type 1 (also called infantile onset or Werdnig-Hoffman disease) is the most prevalent form of the disease. Patients with type 1 generally have 2 copies of SMN2 and present with symptoms before 6 months of age and will not gain the ability to sit. SMA type 2 (also called intermediate SMA or Dubowitz disease) is usually diagnosed after 6 months of age and patients can typically sit but are unable to walk. Patients classified as SMA type 3 (also called juvenile onset or Kugelberg-Welander disease) and SMA type 4 (adult-onset) typically have 3 or more copies of SMN2 and can achieve independent mobility as well as live to adulthood.4,7,13
| Table 1. Classification for Spinal Muscular Atrophy Based on Type4 |
| Spinal Muscular Atrophy Classification |
| Type |
Age at Onset |
Highest Function |
Natural Age at Death |
SMN2 Copies |
| 0 |
Prenatal |
Respiratory support |
<1 month |
1 |
| 1 |
0-6 months |
Never sit |
< 2 years |
2 |
| 2 |
<18 months |
Never stand |
>2 years |
3, 4 |
| 3 |
>18 months – 21 years |
Stand alone |
Adult |
3, 4 |
| 4 |
>21 years |
Stand alone |
Adult |
4-8 |
More recently, as SMA therapies have evolved, patients have experienced increased motor function. This has blurred the lines between previously defined SMA types and shifted the focus to functional status rather than onset of symptoms and SMN2 count. The new classification of SMA divides patients into functional groups such as nonsitters, sitters, and walkers.14
SUPPORTIVE THERAPY
Classification of severity can assist in determining the approach to management for patients with SMA. Historically, SMA therapy consisted of more supportive measures focusing on organs that can be affected by the disease state such as neuromuscular/orthopedic, pulmonary, and gastrointestinal and bone health (nutritional). Neuromuscular and orthopedic supportive measures include mostly physical management through rehabilitation for muscular atrophy with goals based on the patient’s functional classification, as well as thoracic bracing or surgery for spinal malformation.14
Progressive muscular atrophy associated with SMA also has an effect on patients’ respiratory tracts with an increased risk of respiratory infections. Patients use manual, medicinal, and mechanical methods to help prevent these infections. Manual chest physiotherapy combined with mechanical cough assistance may be used to facilitate airway clearance. Medications that help with airway clearance may also be used; these include nebulized bronchodilators, nebulized mucolytics, and glycopyrrolate. It should be noted that nebulized mucolytics, such as hypertonic saline and dornase-α, should not be used long-term as there is little evidence to support their use, and glycopyrrolate should be used with caution to avoid drying secretions and mucus plugs. Other medications include palivizumab in the first 24 months of life to protect against respiratory syncytial virus, influenza vaccine administered after 6 months of age, and antibiotics for respiratory infections. Although manual and pharmaceutical management may be helpful, a more severe diagnosis may require additional support using mechanical ventilation with continuous positive airway pressure. In some patients, a tracheostomy tube may be required if noninvasive positive pressure ventilation fails.8
SMA patients may require feeding tubes, as loss of muscle control can impair their ability to swallow. In addition to difficulty swallowing, loss of muscle control could contribute to gastroesophageal reflux and necessitate use of histamine H2 receptor antagonists or proton pump inhibitors to prevent discomfort. Some patients may also be at risk for being overweight/obese due to lack of movement from muscular atrophy. Nutrition is supported for these patients through dietary monitoring to ensure that calorie intake and calorie expenditure are proportional while also providing the patient with sufficient nutrition. Patients may also require supplementation with vitamin D, calcium, and bisphosphonate to support bone health.8
Pharmacists can play a large role in increasing the quality of life for patients with SMA through supportive care, especially in the areas of respiratory and gastrointestinal/nutritional health. Additionally, as research has progressed for newer medications that help treat the disease state versus its symptoms, the pharmacist’s role has expanded to undertake a new responsibility for managing these emerging treatment options (see Table 2 for a summary of supportive therapy options).
| Table 2. Summary of SMA Supportive Therapy Based on Functional Status8,14 |
| Classification |
Support |
Assessment |
Intervention |
| Nonsitter |
Rehabilitation
|
· Functional scale- CHOP-INTEND
· Motor development-HINE
|
· Positioning and bracing
· Stretching
· Promote function and mobility
|
|
Pulmonary
|
· Physical exam
· Sleep study
· End tidal CO2 for hypoventilation
|
· Airway clearance support
· Respiratory therapy
· Noninvasive ventilation
|
|
Nutritional
|
· Swallow study
· Analysis of nutrition
· Vitamin D levels
· Glucose metabolism
· Constipation
· Gastroesophageal reflux
|
· Ensure adequate fluid/electrolytes
· Provide calcium and vitamin D
· Monitor glucose
· Bowel regimen
|
| Sitter |
Rehabilitation
|
· Functional scales: HFMSE, RULM, MFM
· Muscle weakness: strength tests
|
· Positioning and bracing
· Stretching
· Promote function and mobility
|
|
Pulmonary
|
· Physical examination
· Spirometry (age dependent)
· Sleep study
|
· Nebulized bronchodilators
· Immunizations
· Palivizumab
· Support airway clearance
· Respiratory therapy
· Noninvasive ventilation
|
|
Nutritional
|
· Swallow study
· Symptoms of dysphagia, aspiration, difficulty feeding
· Height, weight
· Nutritional analysis
· Glucose metabolism
· Vitamin D
· Constipation
· Gastroesophageal reflux
|
· Ensure adequate fluid/electrolytes
· Provide calcium and vitamin D
· Monitor glucose
· Bowel regimen
|
| Ambulant |
Rehabilitation
|
· Measure of endurance — 6MWT
· Functional scales — HFMSE, RULM
· Muscle weakness- strength test
|
· Positioning and bracing
· Stretching
· Promote function and mobility
|
|
Pulmonary
|
· Clinical examination
· Review of cough effectiveness
· Monitor cough effectiveness
|
· Nebulized bronchodilators
· Immunizations
· Palivizumab
|
|
Nutritional
|
· Nutritional analysis
· Glucose metabolism
· Vitamin D
|
· Provide calcium and vitamin D
|
| Abbreviations used: CHOP-INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; HFMSE, Hammersmith Functional Motor Scale-Expanded Score; HINE, Hammersmith Infant Neurologic Exam; MFM, Motor Function Measurement; 6MWT, 6-minute walk test; RULM, The Revised Upper Limb Module. |
CURRENT THERAPIES
Beyond supportive care, medications that could alter the disease course of SMA was previously lacking. In 2012, a Cochrane review published 6 randomized placebo-controlled trials on treatment for SMA. These trials used creatine, phenylbutyrate, gabapentin, thyrotropin-releasing hormone, hydroxyurea, and dual therapy with valproate and acetyl-L-carnitine. None of these medications demonstrated significant effects or benefit in SMA patients. 15
Only recently have disease-modifying agents emerged and changed the natural history of SMA. The currently FDA-approved, SMN-directed therapies consist of nusinersen, onasemnogene abeparvovec-xioi, and risdiplam.
Nusinersen
Nusinersen was the first agent on the U.S. market; it was approved by the Food and Drug Administration (FDA) in 2016 for the treatment of SMA in pediatric and adult patients.16 Nusinersen is a SMN2-directed antisense oligonucleotide designed to inhibit splicing factors. This facilitates the integration of exon 7 into SMN2 mRNA and subsequently enhances full-length SMN proteins (see Figure 3).17 Nusinersen is administered by intrathecal injection in a series of 4 loading doses and maintenance dosing of 12 mg. The first three doses are given at 14-day intervals and the fourth is given 30 days after the third dose. Maintenance doses are administered every 4 months thereafter.
Figure 3. Correction of survival motor neuron gene expression by nusinersen
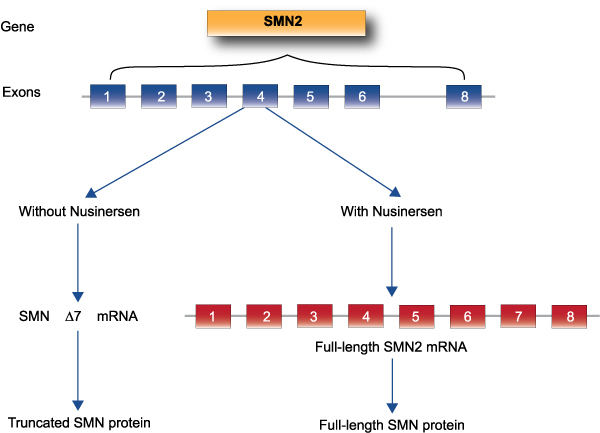
The efficacy of nusinersen was demonstrated in the ENDEAR and CHERISH phase 3 clinical trials.
The ENDEAR trial was a double-blind, sham-controlled study in 121 symptomatic infants ≤7 months of age at the time of the first dose and diagnosed with infantile-onset SMA (type 1). Patients were randomized 2:1 to receive nusinersen or a sham injection. The primary objective for this trial was proportion of responders, which were defined as those with improvements in motor milestones according to Section 2 of the Hammersmith Infant Neurologic Exam (HINE-2). The HINE assessment scores 7 different areas of motor milestone development (maximum score between 2–4 points for each milestone and a total of 26 points). Treatment responder was defined by a 2-point increase in ability to kick, or at least a 1-point increase in head control, rolling, sitting, crawling, standing, or walking. Patients also had to exhibit improvement in more milestones than worsening.
The interim analysis for the ENDEAR trial showed 41% of nusinersen-treated patients (n = 51) achieved a motor milestone response compared with none of the sham-controlled patients (P <0.001).19 This prompted early termination of the trial, and patients were enrolled in an ongoing extension trial, SHINE. The final analysis of ENDEAR showed a significantly higher percentage of infants with improvements in motor-milestone response in the nusinersen group (51%) versus the control group (37%). The authors also reported the nusinersen group had a higher likelihood of event-free survival (HR, 0.53; P = 0.005) and overall survival (HR, 0.37; P = 0.004).19
CHERISH was a similarly designed phase 3 trial evaluating nusinersen in patients aged 2 to 12 years old diagnosed with later-onset SMA (likely type 2 or 3). The primary endpoint was improvement in motor function assessed using the Hammersmith Functional Motor Scale-Expanded (HFMSE) score. As in the ENDEAR trial, patients receiving nusinersen had clinically significant improvements during an interim analysis, which prompted early termination of the trial.20
Other notable ongoing nusinersen studies include NURTURE and DEVOTE. The NURTURE trial is a phase 2 study evaluating the initiation of nusinersen therapy in infants during the presymptomatic stage of SMA. Early intervention, in this case before presentation of symptoms, can prevent the irreversible loss of motor neurons. This ongoing study is a multisite, open-label, single-arm trial in 25 children with genetically diagnosed SMA who first received nusinersen in infancy (3–42 days old). Of the 25 patients, 15 have 2 copies of the SMN2 gene and 10 have 3 SMN2 copies. The primary objective of this study is time to death or respiratory intervention (invasive or noninvasive for ≥6 hours per day continuously for ≥7 days or tracheostomy). The secondary objectives include proportion of participants alive; attainment of motor milestones as assessed by World Health Organization criteria, attainment of motor milestones by HINE-2, change from baseline in the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) scale, change from baseline in growth parameters, and proportion of participants developing clinically manifested SMA at 13 and 24 months of age.
In the interim analysis of the NURTURE study, all patients were alive and past the expected age of symptom onset. A total of 4 infants were using respiratory interventions for 6 or more hours per day continuously for 7 or more days due to acute, reversible illness. All 25 participants achieved the ability to sit without support, 92% achieved walking with assistance, and 88% achieved walking independently.21 The NURTURE study’s interim analysis concluded that nusinersen demonstrated a favorable risk-benefit profile consistent with data from previous studies.
The DEVOTE study, which began in 2020, is a randomized, double-blind, dose finding trial with the primary objective of examining the clinical efficacy of intrathecal nusinersen administration at higher doses. The secondary objectives of this trial include assessments of safety, tolerability, and pharmacokinetics of high dose nusinersen.21 See Table 3 for a summary of nusinersen trials.
| Table 3. Summary of Nusinersen Trials17,19–22 |
| Trial |
Phase |
Method |
Population |
Results |
|
ENDEAR
|
3
|
Multicenter, randomized, double-blind, sham-controlled
Nusinersen dose: 12 mg (or equivalent) on days 1, 15, 29, 64, 183, and 302
Primary endpoint:
· Proportion of responders (measured by HINE-2 scores)
|
121 infants
≤7 months of age with early-onset SMA (type 1)
|
· Improvement of motor milestone: nusinersen 51% vs sham 0%, P <0.001
· Likelihood of event-free survival: HR 0.53; P = 0.005
· Likelihood of overall survival: HR 0.37; P = 0.005
|
|
CHERISH
|
3
|
Multicenter, randomized, double-blind, sham-controlled
Nusinersen dose: 12 mg on days 1, 29, 85, and 274
Primary endpoint:
· Change in HFMSE
|
126 children
2–12 years of age with
later-onset SMA (types 2 or 3)
|
· Change in HMFSE (increase ≥3 points from baseline): nusinersen 57% vs control group 26% increase
|
|
NURTURE
|
2
|
Multisite, open-label, single-arm
Nusinersen dose: 12 mg on days 1, 15, 29, 64, then every 119 days
Primary endpoint:
· Death
· Time to respiratory intervention
|
25 infants
3–42 days old with
genetically diagnosed SMA
|
Ongoing
|
|
SHINE
|
3
|
Nonrandomized, parallel assignment, triple-blind
Primary objective:
· Long-term safety and tolerability of intrathecal nusinersen in patient who previously participated in investigational studies of nusinersen
|
292 patients
pediatric and adult from previous studies
|
Ongoing
Interim:
Stable HFMSE and RULM scores
|
|
DEVOTE
|
2/3
|
Randomized, double-blind, dose-finding trial
Nusinersen dose (load/maintenance): 28/28 mg, 12/12 mg, 50/28 mg and previous 12 mg followed by 50/28 mg
Primary objective:
· Clinical efficacy of intrathecal nusinersen administration at higher doses
|
125 patients of all ages
|
Ongoing
|
| Abbreviations used: HFMSE, Hammersmith Functional Motor Scale-Expanded Score; RULM, The Revised Upper Limb Module; SMA, spinal muscular atrophy. |
In the CHERISH study, adverse events such as lower and upper respiratory tract infection and constipation were the most commonly seen in patients who received nusinersen. The ENDEAR study showed no meaningful differences in adverse events between the nusinersen and sham groups. However, the most notable adverse events in these groups were pyrexia, constipation, respiratory distress, and upper respiratory tract infections. The NURTURE trial showed fevers, upper respiratory tract infections, cough, nasopharyngitis, and vomiting to be the most prevalent adverse events for patients using nusinersen.19,20
Nusinersen has a label warning for thrombocytopenia and coagulation abnormalities as well as renal toxicity. In both the ENDEAR and CHERISH studies, thrombocytopenia was higher among nusinersen-treated patients compared with sham-controlled patients. The risk of thrombocytopenia and coagulation abnormalities may increase patient risk of bleeding.19 Also, in the ENDEAR and CHERISH studies, 71 (58%) of nusinersen-treated patients had elevated urine protein compared with 22 (34%) of sham-controlled patients. There were no elevations in serum creatinine or cystatin C observed. Because of these warnings, laboratory tests at baseline for patients treated with nusinersen include platelet count, prothrombin time, activated partial thromboplastin time, and urine protein.17
Onasemnogene Abeparvovec-xioi
In 2019, the FDA approved a second medication that had showed efficacy in the treatment of SMA. Onasemnogene abeparvovec-xioi is a gene therapy approved for the treatment of pediatric SMA patients younger than 2 years of age with bi-allelic mutations in the SMN1 gene.23 Onasemnogene is an adeno-associated virus vector-based gene therapy, AAV9-SMN1, is designed to deliver a copy of the gene encoding the human SMN protein. This results in transgene expression in the brain, spinal motor neurons, dorsal root ganglia neurons, and glial cells throughout the central nervous system in addition to widespread expression in the liver, heart, and skeletal muscle.24 Unlike nusinersen, onasemnogene is a one-time intravenous (IV) infusion for the treatment of SMA.
The safety and efficacy of this SMN1 gene therapy was supported by a phase 1 study evaluating 15 patients with infantile-onset SMA (type 1) with 2 copies of SMN2 who received a single dose of onasemnogene. Three patients received a low dose (6.7 x 1013 vg/kg) and 12 patients received a high dose (2 x 1014 vg/kg) IV infusion. After receiving the gene therapy, the first patient had elevated serum aminotransferase levels and was treated with prednisolone. Because of this, subsequent patients were administered prednisolone 1 mg/kg daily for approximately 30 days, starting 1 day before viral delivery in order to suppress the immune response to a high viral load. The primary outcome was safety and the secondary outcome was time until death or the need for permanent ventilator assistance. In a further exploratory analysis, the CHOP-INTEND scale was used to compare scores of the 2 cohorts. The CHOP-INTEND assessment uses a 0 to 63 point scale in which higher scores indicate better motor function.25
At the conclusion of the study, all 15 patients were alive and event-free at 20 months of age, as compared with a rate of survival of 8% in a historical cohort. There was significant increase from baseline in the CHOP-INTEND score in the high-dose cohort following gene delivery (9.8-point increase at 1 month, 15.4-point increase at 3 months). Of the 12 patients who received high-dose onasemnogene, 11 were able to sit unassisted, 9 were able to roll over, 11 could eat orally and speak, and 2 walked independently. The most common side effects included elevated serum aminotransferase levels, which was mitigated by prednisolone. This study concluded that in patients with SMA type 1, a single dose of onasemnogene resulted in longer survival, superior achievement of motor milestones, and better motor function when compared to historical cohorts, though the authors noted the need for further studies to confirm these findings.26
A follow-up phase 3, open-label, single-arm, single-dose pivotal trial (STR1VE) has been completed to evaluate the efficacy of onasemnogene in patients with SMA type 1 and 1 or 2 SMN2 copies who were younger than 6 months of age.24,27 The primary outcome of this study was the achievement of independent sitting for at least 30 seconds (at 18 months of age) and event-free survival. Secondary outcomes included the ability to thrive, defined as the ability to tolerate thin liquids through a formal swallowing test, not requiring mechanical support or other nonoral method, and able to maintain weight as well as independence from ventilator support by the time the patient was 18 months old.27 Preliminary results from STR1VE showed prolonged event-free survival, increased CHOP-INTEND score, and milestone achievement in SMA patients treated with onasemnogene.28
In addition to these trials, other ongoing studies are assessing use onasemnogene in patients with SMA (see Table 4). SPR1NT is an ongoing phase 3, open-label, single-arm, single-dose trial evaluating presymptomatic patients younger than 6 months of age with 2 to 3 copies of SMN2 treated with onasemnogene.29 Preliminary data from SPR1NT showed patients achieved age-appropriate motor milestones. An ongoing, long-term, 15-year, safety study, START, is following 10 patients from the high-dose cohort from the initial phase 1 trial.28
Onasemnogene has been a relatively well-tolerated drug. The most common adverse reactions have been elevated aminotransferases and vomiting. Combining results from 4 open-label studies conducted in the United States, a total of 44 patients with SMA received onasemnogene. Of these, 41 patients received doses at or above the recommended dose and 3 at a lower dose. From this patient population, 12 (27.3%) patients had elevated transaminases and 3 (6.8%) patients experienced vomiting. One patient in clinical trials experienced hepatic dysfunction with jaundice and increased aspartate aminotransferase (AST), alanine aminotransferase (ALT), bilirubin, and prothrombin time. This patient recovered to baseline after treatment with corticosteroids. One patient experienced respiratory insufficiency 12 days after a dose, but this was caused by respiratory syncytial virus and parainfluenza from respiratory secretions.30
The pharmacokinetics for onasemnogene abeparvovec-xioi has been investigated thoroughly. Samples of saliva, urine, and stool were collected the day after the infusion, weekly through day 30, monthly until month 12, with subsequent samples every 3 months thereafter until month 18 and analyzed for vector shedding based on vector DNA concentrations. The highest vector DNA concentration was found in stool, where levels were higher than in saliva or urine for 1–2 weeks after infusion and declined to undetectable levels after 1–2 months. In saliva, vector DNA concentrations were low on day 1 after infusion and declined to undetectable by week 3. Vector DNA concentrations in urine were very low on day 1 and declined to undetectable levels within 1–2 weeks.30
Biodistribution was also evaluated in patients who had received the recommended dosing and had died. It was found that vector DNA was more commonly found in the liver, but also reached the spleen, heart, pancreas, inguinal lymph node, skeletal muscles, peripheral nerves, kidney, lung, intestines, spinal cord, brain, and thymus.30
| Table 4. Summary of Onasemnogene Trials24–29 |
| Trial |
Phase |
Method |
Population |
Results |
|
Single-Dose Gene-Replacement Therapy for SMA
|
1
|
Dose-finding, safety trial
Dose: Low dose (6.7 x 1013 vg/kg), high dose (2 x 1014 vg/kg)
Primary outcome:
· Safety
Secondary outcome:
· Time until death or need for permanent ventilation
· Assessment of motor function with CHOP-INTEND
|
15 patients
Infantile-onset SMA (type 1) with 2 copies of SMN2
|
Alive and event free at 20 months: 100%
Increase in CHOP-INTEND score (high dose):
· 1 month: 9.8 points
· 3 months: 15.4 points
Adverse Effects: Elevated serum aminotransferase levels
|
|
STR1VE
|
3
|
Open-label, single-arm, single-dose trial
Primary outcome (measured at 18 months):
· Achievement of independent sitting for at least 30 seconds
· Event free survival
Secondary outcome (measured at 18 months):
· Ability to thrive (tolerates thin liquids, does not require mechanical support, able to maintain weight)
|
22 patients
Infantile-onset SMA (type 1) with 1 to 2 copies of SMN2
|
Preliminary Results
· 95% event-free survival without the need for permanent ventilation
· CHOP-INTEND
o 1 month: +6.9 points
o 3 months: +11.7 points
o 5 months: +14.3 points
o Score ≥40: 21 patients
· BSID-III Criteria
o Sit 30 seconds without support: 11 patients
|
|
SPR1NT
|
3
|
Open-label, single-arm, single-dose trial
Primary objective:
· Efficacy and Safety
|
29 patients
Presymptomatic patients ≤ 6 weeks old with
2 to 3 copies of SMN2
|
Preliminary Results
· 2 copies of SMN2
o Ability to sit independently: 57%
o Ability to walk independently: 28.5%
· 3 copies of SMN2
o Ability to stand alone: 26.6%
|
|
START
|
n/a
|
Observational, long-term, follow-up study
Primary objective:
· Efficacy and Safety
|
10 patients
Infantile-onset SMA (type 1)
|
Preliminary Results
All patients are alive and free of permanent ventilation
|
| Abbreviations used: BSID-III, Bayley Scales of Infant and Toddler Development, third edition; CHOP-INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; SMA, spinal muscular atrophy; SMN, survival motor neuron. |
Risdiplam
The agent most recently approved by FDA for treatment of SMA in patients 2 months of age and older is risdiplam. This is a small-molecule splicing modifier that promotes the inclusion of exon 7 in SMN2 mRNA and the production of full-length functional SMN protein. Risdiplam is also novel as it is a daily, orally administered, liquid medication.31
There are 4 notable studies evaluating safety and efficacy of risdiplam in SMA: FIREFISH, SUNFISH, JEWELFISH, and RAINBOWFISH. These are summarized in Table 5 and discussed below. Some data were supplied by the company marketing risdiplam.
FIREFISH is an ongoing, open-label, 2-part clinical trial in infants aged 1 to 7 months with infantile-onset SMA type 1. Part 1 was a dose-finding study in 21 infants with the primary objective of assessing safety, tolerability, pharmacokinetics, and pharmacodynamics of risdiplam. Part 2 of FIREFISH includes 41 infants and will assess safety and efficacy of the dose selected in part 1 for 24 months followed by an open-label extension phase. The primary outcome measure is the proportion of infants sitting without support for at least 5 seconds by 12 months of treatment as measured by the Gross Motor Scale of the Bayley Scales of Infant and Toddler Development, third edition (BSID-III). Findings at 12 months show 29% of patients met the primary endpoint (P <0.0001), and 56% of infants achieved a CHOP-INTEND total score of 40 or more. The most common adverse effects were upper respiratory tract infection, pneumonia, pyrexia, constipation, nasopharyngitis, rhinitis, and diarrhea.31,32
SUNFISH is also a phase 2/3, double-blind and placebo-controlled, 2-part study. It includes patients aged 2–25 years with later-onset SMA types 2 and 3. Part 1 included 51 patients and had similar design to FIREFISH. Part 2 evaluated the efficacy and safety of risdiplam with the primary objective of showing a significant mean change from baseline at 12 months in the total Motor Function Measure 32 (MFM-32) score compared with placebo. SUNFISH part 2 also met its primary endpoint and adverse events were similar to those of the FIREFISH trial (data supplied by manufacturer).
JEWELFISH is an open-label exploratory trial in patients with SMA aged 6 months to 60 years previously treated with SMA-directed therapies. RAINBOWFISH is an open-label, single-arm, multicenter study, investigating the efficacy, safety, pharmacokinetics, and pharmacodynamics of risdiplam in presymptomatic infants from birth to 6 weeks of age with genetically diagnosed SMA (data supplied by manufacturer).
| Table 5. Summary of Risdiplam Trials |
| Trial |
Phase |
Method |
Population |
Results |
| FIREFISH |
2/3
|
Open-label, two-part study
Part 1 primary objective: safety and dose finding
· The motor function assessed through CHOP-INTEND
Part 2 primary objective: efficacy
· BSID-III assessment of sitting without support
|
Part 1: 21 infants
Infantile-onset SMA type 1
· Low-dose cohort: 4
· High-dose cohort: 17
Part 2: 41 infants
Treated for 24 months
|
Part 1
· CHOP-INTEND ≥40: 82% of high-dose group
· Event-free survival: 85.7%
Part 2
· BSID-III: 29% met criteria
· HINE-2: 78% met criteria
|
| SUNFISH |
2/3
|
Double-blind, placebo-controlled, multicenter
Part 1 objectives:
· Safety and tolerability
· Pharmacokinetics and pharmacodynamics
· Dose finding
Part 2 primary endpoint: efficacy and safety
· Mean change in the MFM-32 total score at 12 months compared with placebo
|
Part 1: 51 patients
2–25 years old
Later-onset SMA types 2 or 3
Part 2: 180 patients
|
Part 1
· Median 2-fold increase in SMN protein after 4 weeks of therapy
· MFM-32 score increased by average of 2.66
Part 2
MFM-32 score mean change
· Risdiplam: +1.36 (95% CI 0.61, 2.11)
· Placebo: –0.19 (95% CI: –1.22, 0.84)
|
| JEWELFISH |
2
|
Open-label, exploratory study
Primary objective: safety, tolerability, pharmacokinetics, and pharmacodynamics
|
Patients aged 6 months to 60 years with SMA
Previously treated with other SMA-directed therapies
|
Ongoing
|
| RAINBOW FISH |
2
|
Open-label, single-arm, multicenter study
Primary objective:
· Efficacy and safety
· Pharmacokinetics and pharmacodynamics
|
Patients from birth to 6 weeks of age
Presymptomatic
|
Ongoing
|
Source: Some data supplied by the company marketing risdiplam.
Abbreviations used: BSID-III, Bayley Scales of Infant and Toddler Development — third edition; CHOP-INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; HINE-2, Hammersmith Infant Neurologic Exam 2; MFM, Motor Function Measurement; SMA, spinal muscular atrophy; SMN, survival motor neuron. |
EMERGING THERAPIES
Currently, nusinersen, onasemnogene abeparvovec-xioi, and risdiplam are the only FDA-approved treatment options for patients with SMA; however, new therapies in various stages of development may further expand the treatment landscape for patients with SMA. Branaplam, reldesemtiv, and SRK-015 will be discussed in this program.
Branaplam
Branaplam is an oral splicing modifier similar to risdiplam. An ongoing multipart, open-label, first-in-human study will investigate once-weekly branaplam in patients with SMA. Initial enrollment was halted because of safety concerns when animal trials showed injuries to peripheral nerves, spinal cord, testes, and blood vessels in the kidney. The study has resumed and completed enrollment.33
SRK-015
Another agent currently under study is a fully human monoclonal antibody, SRK-015. This drug selectively binds and inhibits the latent, or inactive form, of myostatin found in skeletal muscles. Myostatin is part of the TGFβ superfamily of growth factors and inhibits muscle cell growth and differentiation. Although this mechanism does not target the genetic cause of SMA directly, SMN-independent therapies such as SRK-015 may help to improve muscle mass and function.
The first placebo-controlled, double-blind, phase 1 clinical trial for SRK-015 was completed in 2018 and supported the tolerability of SRK-015 in doses up to 30 mg/kg, with no dose-limiting toxicities reported. A phase 2 trial, TOPAZ, was initiated to study the safety and efficacy of SRK-015 in patients aged 2–21 years old with later-onset SMA (types 2 and 3). The primary outcome measure in this trial is change in motor function or physical abilities based on the Revised Hammersmith Scale (RHS) and the HFMSE. This trial is expected to be completed in 2021.24
Reldesemtiv
Reldesemtiv, also known as CK-2127107 or CK-107, is another muscle-enhancing therapy, but instead of binding to inactive myostatin, it is a troponin complex activator. By slowing calcium release from fast skeletal muscle troponin, reldesemtiv sensitizes the sarcomere to calcium, resulting in increased motor nerve stimulation. Preclinical trials in rats showed increased stamina in skeletal muscle in vivo. A phase 2, double blind, randomized, multidose study in patients 12 years or older with SMA types 2, 3, and 4 evaluated the pharmacodynamic effect of reldesemtiv versus placebo. The outcome of this trial is pending.24
Reldesemtiv does not work on chromosome 5, is not supportive care, and does not cure SMA.
Pharmacy Considerations
With new evolving therapies in a complex disease, it is important for pharmacists to be knowledgeable about the unique mechanism of each drug to optimize pharmaceutical care for SMA patients. Pharmacists have a role in education for patients, caregivers, and providers as well as in formulary management and creating guidelines surrounding proper storage, preparation, dispensing, and administration of novel drugs such as onasemnogene.
MANAGEMENT OF ADVERSE EFFECTS
Therapies that have been approved for SMA, while effective and groundbreaking, are not without risk. Serious adverse reactions associated with the earliest approved SMA agent, nusinersen, include thrombocytopenia, coagulation abnormalities, and renal toxicities. Platelet count, coagulation tests, and spot urine protein levels should be monitored at baseline and prior to every administration of nusinersen.16
The most serious adverse events associated with onasemnogene include elevated aminotransferases, acute liver injury, and thrombocytopenia. Liver function tests — including AST/ALT, total bilirubin, and prothrombin time — should be monitored weekly for the first month, every other week for the second and third months and until results are unremarkable. In the United States, product labeling for onasemnogene has a boxed warning for acute serious liver injury, and patients with pre-existing liver impairment may be at increased risk. One patient treated under the expanded access program had elevated aminotransferase levels before administration of onasemnogene, received corticosteroids for 30 days, and developed acute serious liver injury about 7 weeks after administration. Additionally, 2 patients developed elevated aminotransferases after onasemnogene in clinical trials. All patients recovered with treatment of corticosteroids. Oral corticosteroids equivalent to prednisolone 1 mg/kg/day should be started 1 day before onasemnogene infusion and continued for 30 days to mitigate risk of elevated aminotransferases. Thrombocytopenia should be monitored by platelet counts at baseline, weekly for the first month, and then every other week for the second and third months until return to baseline. Transient elevated troponin-1 levels were also observed in clinical trials and should be monitored before and following infusion.30
Adverse reactions associated with the most recently marketed SMA agent, risdiplam, consist of fever, diarrhea, and rash in later-onset SMA and upper respiratory tract infection, pneumonia, constipation, and vomiting in early-onset SMA patients. These adverse effects are managed symptomatically.31
PARENT/CAREGIVER COUNSELING
Upon initiation of SMA therapy, caregivers can benefit from medication counseling by a pharmacist. For patients on nusinersen therapy, it is recommended to inform patients and caregivers that nusinersen may increase the risk of bleeding or cause renal toxicity. It is also important to note that they should seek medical attention if unexpected bleeding occurs.
Onasemnogene counseling should include risk of increased liver enzymes and acute serious liver injury and the importance of receiving oral corticosteroid medication before and after infusion to mitigate these adverse effects. Caregivers should be advised to contact their healthcare provider if the patient’s skin and/or whites of eyes become yellowish. Counseling should also include risk of bruising and bleeding as well as when to seek medication attention. Temporary vector shedding after onasemnogene infusion occurs through body waste, and caregivers should be aware of precautions to protect themselves and others from exposure. Proper handling of stool, including using disposable diapers, sealing diapers in disposable trash bags, and proper hand hygiene should be followed for 1 month after onasemnogene infusion.30
Counseling for risdiplam therapy includes pregnancy and fetal risk, potential effects on male fertility, and instructions for the oral solution preparation. In animal studies, risdiplam was noted to potentially cause fetal harm. It is recommended to advise women with childbearing potential to use effective contraception during treatment with risdiplam and for at least 1 month after therapy. Risdiplam therapy is available as an oral solution, and pharmacists should counsel on proper storage as well as instructions for administration. Patients and caregivers should be counseled to take this product after a meal or after breastfeeding at approximately the same time each day. This medication should not be mixed with formula or milk.31
PROVIDER CONSULTATION
In addition to monitoring of potential adverse effects previously discussed, pharmacists must work with frontline clinicians on procedures for ordering, handling, and administering novel SMA therapies. Nusinersen should be administered at room temperature within 4 hours of drawing up a patient-specific dose. Before administration, 5 mL of cerebrospinal fluid should be removed and nusinersen administered by intrathecal injection.16
Onasemnogene is dosed in vector genomes per kilogram of body weight. Dosing is specific to a weight range and corresponding volume (refer to prescribing information). Before onasemnogene infusion, baseline anti-AAV9 antibody testing should be performed in addition to previously mentioned laboratory tests for monitoring of adverse events. Pharmacists can also counsel providers on how to adjust vaccination schedules if needed to accommodate treatment with corticosteroids and discuss prophylaxis against respiratory syncytial virus for patients up to 2 years of age. Onasemnogene should be administered as slow infusion over 60 minutes and should not be administered as a push or bolus. A back-up catheter should be inserted in case the primary catheter becomes nonfunctional.30
Since risdiplam is orally administrated, pharmacists can educate providers on how to counsel patients on dosing and administration. Dose is determined by patient age and weight. Missed doses of risdiplam should be administered as soon as possible if within 6 hours of missed dose and the usual dose can be resumed the next day. If outside the 6-hour window, the dose should be skipped and the regularly scheduled dose resumed the next day. If risdiplam is not fully swallowed or if the patient vomits the dose, another dose should be administered.31
FORMULARY DEVELOPMENT
Before instituting new medications such as gene therapy within the health system, many facets of storage and handling, preparation, administration, and disposal should be considered. Protocols will need to be implemented to ensure therapies are ordered and administered safely. Without specific guidance from regulatory bodies, institutions will have to address these concerns with their own policies and procedures. A multidisciplinary team comprising pharmacists, physicians, nurses, administrative leaders, environmental and medication safety officers, and members of finance teams may be needed.34 Pharmacists, both administrative and clinical, will need to be involved in creating institutional policies and procedures.
For gene therapy or hazardous medications, such as onasemnogene, pharmacists who are experts in sterile compounding will need to develop standard operating procedures on how to handle and prepare these products and minimize environmental or staff exposure. Biosafety guidelines from the National Institutes of Health (NIH), Biosafety in Microbiological and Biomedical Laboratories (BMBL), and the National Institute for Occupational Safety and Health (NIOSH) may be helpful to guide policy decisions. For safe handling and compounding of sterile medications, the United States Pharmacopeia (USP) General Chapters <797> and <800> should be followed. USP <797> and <800> outline specific procedures for handling sterile and hazardous medications and take into account exposure risk, route of administration, manipulation requirements, and packaging. These guidelines also provide guidance for disposal, decontamination, cleaning, and spills.34
Recently, a pharmacist-driven review of gene replacement therapy was published that offers guidance to health-system pharmacists. This article recommends having institutional procedures with training for personnel and education for patient and caregivers and pharmacy-driven policy and procedures for medication handling and administration as well as environmental controls to minimize risk of harm.35
Clinical pharmacists are also important for formulary development by leading discussions on indications and restrictions, clinical pathway development, and medication counseling. They can also work with providers on management of electronic health record related to medication ordering and medication administration. Pharmacists in specialty and managed care pharmacies will also have a critical role in working with providers, patients, and payers on the availability and affordability of these new drugs.
CONCLUSION
SMA is a complex disease that stems from a mutation in the SMN1 gene, causing irreversible loss of motor neuron cells. SMA leads to progressive muscle weakness and respiratory, cardiac, and gastrointestinal complications. SMA management previously consisted of supportive care. However, recent novel treatments can prevent the progression of muscle degradation; these have changed the course of SMA management. Three agents have been approved by FDA for the treatment of SMA: nusinersen, onasemnogene abeparvovec-xioi, and risdiplam. As these agents are becoming more accessible to patients, it is important for pharmacists to be aware of possible adverse effects and logistical information to facilitate proper medication administration to the patient.
In addition to the FDA-approved therapy, new agents are in development that could modify available options for care of patients with SMA. Future trials of these agents will assess their safety, efficacy, and place in therapy, as well as their use in combination with currently approved medications. As these trials progress, it will be important for pharmacists to stay informed and educated about this important condition and ways of managing its detrimental effects.
- Pearn J. Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet. 1978;15(6):409–413.
- Verhaart IEC, Robertson A, Wilson IJ, et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy — a literature review. Orphanet J Rare Dis. 2017;12(1):124.
- Belter L, Cook SF, Crawford TO, et al. An overview of the Cure SMA membership database: highlights of key demographic and clinical characteristics of SMA members. J Neuromuscul Dis. 2018;5(2):167–176.
- Kolb SJ, Kissel JT. Spinal muscular atrophy. Neurol Clin. 2015;33(4):831–846.
- Gubitz AK, Feng W, Dreyfuss G. The SMN complex. Exp Cell Res. 2004;296(1):51–56.
- Gendron NH, MacKenzie AE. Spinal muscular atrophy: molecular pathophysiology. Curr Opin Neurol. 1999;12(2):137–142.
- Arnold WD, Kassar D, Kissel JT. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve. 2015;51(2):157–167.
- Finkel RS, Mercuri E, Meyer OH, et al. Diagnosis and management of spinal muscular atrophy. Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. 2018;28(3):197–207.
- Hendrickson BC, Donohoe C, Akmaev VR, et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J Med Genet. 2009;46(9):641–644.
- Fallon L, Harton GL, Sisson ME, et al. Preimplantation genetic diagnosis for spinal muscular atrophy type I. 1999;53(5):1087.
- Kay DM, Stevens CF, Parker A, et al. Implementation of population-based newborn screening reveals low incidence of spinal muscular atrophy. Genet Med. 2020;22:1296–1302.
- Newborn Screening for Spinal Muscular Atrophy — Cure SMA. Cure SMA website. 2020. Accessed September 1, 2020. www.curesma.org/newborn-screening-for-sma/
- Lally C, Jones C, Farwell W, Reyna SP, Cook SF, Flanders WD. Indirect estimation of the prevalence of spinal muscular atrophy type I, II, and III in the United States. Orphanet J Rare Dis. 2017;12(1):175.
- Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28(2):103–115.
- Bosboom WMJ, Vrancken AFJE, van den Berg LH, Wokke JHJ, Iannaccone ST. Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst Rev. 2009(1):CD006282. doi: 10.1002/14651858.CD006282.pub2
- Spinraza prescribing information. Biogen, Inc. website. 2019. Accessed August 17, 2020. spinraza-hcp.com/content/dam/commercial/spinraza/hcp/en_us/pdf/spinraza-prescribing-information.pdf
- Wurster CD, Ludolph AC. Nusinersen for spinal muscular atrophy. Ther Adv Neurol Disord. 2018;11:1756285618754459.
- Bennett C. ASO therapy for SMA. National Academies website. 2019. Accessed September 1, 2020. https://www.nationalacademies.org/event/04-23 -2019/docs/DEC9D4BA0C5A4AF3F52BAD398FF4739F389C80A77879
- Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723–1732.
- Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378(7):625–635.
- De vivo DC, Bertini E, Swoboda KJ, et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul Disord. 2019;29(11):842–856.
- Study of nusinersen (BIIB058) in participants with spinal muscular atrophy (DEVOTE). ClinicalTrials.gov website. January 27, 2020. Accessed August 23, 2020. clinicaltrials.gov/ct2/show/NCT02462759
- Jędrzejowska M, Borkowska J, Zimowski J, et al. Unaffected patients with a homozygous absence of the SMN Eur J Hum Genet. 2008;16:930–934.
- Shorrock HK, Gillingwater TH, Groen EJN. Overview of current drugs and molecules in development for spinal muscular atrophy therapy. 2018;78(3):293–305.
- Glanzman AM, Mazzone E, Main M, et al. The Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): test development and reliability. Neuromuscul Disord. 2010;20(3):155–161.
- Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713–1722.
- Gene replacement therapy clinical trial for patients with spinal muscular atrophy type 1 (STR1VE). ClinicalTrials.gov website. July 16, 2020. Accessed August 23, 2020. clinicaltrials.gov/ct2/show/NCT03306277
- Zolgensma data shows rapid, significant, clinically meaningful benefit in SMA including prolonged event-free survival, motor milestone achievement and durability now up to 5 years post-dosing [news release]. Novartis website. March 24, 2020. Accessed April 2, 2020. novartis.com/news/media-releases/zolgensma-data-shows-rapid-significant-clinically-meaningful-benefit-smaincluding-prolonged-event-free-survival-motor-milestone-achievement-and-durability-now
- Pre-symptomatic study of intravenous onasemnogene abeparvovec-xioi in spinal muscular atrophy (SMA) for patients with multiple copies of SM2 (SPR1NT). ClinicalTrials.gov. May 18, 2020. Accessed September 1, 2020. clinicaltrials.gov/ct2/show/NCT03505099
- Zolgensma prescribing information. Avexis, Inc. website. 2020. Accessed September 1, 2020. zolgensma.com/?gclid=EAIaIQobChMI1oHnkM7J6wIVBI_ICh2W9gwREAAYASAAEgL3e_D_BwE
- Evrydsi prescribing information. Genentech USA website. 2020. Accessed September 1, 2020. www.gene.com/download/pdf/evrysdi_prescribing.pdf
- Baranello G et al. FIREFISH Part 1: 1-year results on motor function in babies with type 1 SMA. S25.003. Presented at 71st American Academy of Neurology Annual Meeting, Philadelphia, Pennsylvania, May 4–10, 2019.
- An open label study of LM1070 (Branaplam) in type 1 spinal muscular atrophy (SMA). ClinicalTrials.gov website. July 28, 2020. Accessed September 1, 2020. clinicaltrials.gov/ct2/show/NCT02268552
- Beans BE. USP <800> adds significant safety standards: facility upgrades needed to protect employees from hazardous drugs. P T. 2017;42(5):336–339.
- Petrich J, Marchese D, Jenkins C, Storey M, Blind J. Gene replacement therapy: a primer for the health-system pharmacist. J Pharm Pract. 2019:897190019854962.
Back to Top