
Expired activity
Please go to the PowerPak
homepage and select a course.
INTRODUCTION
Myelodysplastic syndromes (MDS) are a heterogeneous group of chronic myeloid neoplasms stemming from ineffective clonal hematopoiesis.1 The blood cells are morphologically dysplastic and result in peripheral blood cytopenias and functional abnormalities.2 The clinical course of MDS ranges from a slowly progressive, indolent disease to a more aggressive course that rapidly progresses into acute myeloid leukemia. Ambiguous diagnostic and classification criteria, incomplete evaluation of older patients with cytopenias, and delay in classification of MDS as a cancer until 2001 contribute to inaccurate estimates of the incidence and prevalence of MDS.3,4
The incidence of MDS is approximately 13,400 new cases annually based on data from the Surveillance, Epidemiology, and End Results, or SEER, program, strikingly similar to the incidence of acute myeloid leukemia.4 Analyses of claims data indicate the incidence of MDS may be several times higher, and the prevalence in the United States is an estimated 60,000 to 170,000.5 MDS primarily affect older adults with a median age of 77 years at diagnosis and a slightly male predominance.4 The median overall survival (OS) of patients with MDS is approximately 2.5 years.6
This program reviews currently available and emerging treatments for management of anemia in patients with lower-risk MDS, including ensuring access to patient-specific treatments, toxicity prevention and management, and enhanced pharmaceutical care.
CLINICAL PRESENTATION
Patients with MDS may develop isolated anemia (hemoglobin less than 10 g/dL), neutropenia (less than 1.8 x 109/L), or thrombocytopenia (less than 100 x 109/L) or multiple peripheral cytopenias.1 Some patients are asymptomatic, with 1 or more cytopenias discovered on complete blood count. Patients with anemia may report fatigue, lethargy, malaise, palpitations, dyspnea on exertion, exercise intolerance, or other symptoms associated with hypoxia secondary to anemia.7 Patients with neutropenia may present with infection, and those with thrombocytopenia may present with easy bruising or bleeding.
EVALUATION AND DIAGNOSIS
Diagnosis of MDS is based on morphological assessment of blood and bone marrow, standard metaphase bone marrow cytogenetics, and analysis of acquired genetic mutations in the bone marrow in addition to consideration of duration of cytopenias and other potential causes of cytopenias (i.e., medications, viral infections, or other causes of blood loss such as gastrointestinal bleeding).2,8-10 Minimal diagnostic criteria for MDS includes presence of cytopenia(s) for at least 6 months (or 2 months if accompanied by a specific karyotype associated with MDS or bilineage dysplasia), exclusion of other causes of cytopenia or dysplasia and one of the following: (1) dysplasia of at least 10% in 1 or more of 3 major bone marrow lineages, (2) blast cell count of 5% to 19%, or (3) specific MDS-associated karyotype [e.g., del(5q)].1,10
In the past decade, next-generation sequencing has revealed the genetic diversity in mutations that lead to development of MDS.11 As many as 90% of patients with MDS have at least 1 somatic gene mutation.12 Genetic mutations may confer information about prognosis, facilitate confirmation of MDS diagnosis, guide therapy, or facilitate monitoring of minimal residual disease.13 Mutations are most often found in genes involved in DNA methylation, histone modification, spliceosome machinery, and genes that regulate cell signaling. Some genetic mutations (e.g., TP53, EZH2, ETV6, RUNX1, and ASXL1) negatively affect prognosis and some (e.g., SF3B1) confer a better prognosis in patients with MDS.12,14 Identification of patients harboring del(5q) or an SF3B1 mutation is critical, as patients can be directed toward treatments they are more likely to respond to (i.e., lenalidomide for del(5q) and luspatercept for MDS with ring sideroblasts (MDS-RS) with an SF3B1 mutation). Patients with TET2 and DNMT3A mutations may have a better response with hypomethylating agents (HMAs).12
A subset of patients may not meet diagnostic criteria for MDS, instead harboring one or more somatic mutations with or without cytopenias.15 These patients are part of MDS precursor states known as clonal hematopoiesis of indeterminate potential and clonal cytopenias of unknown significance. While this remains a rapidly growing field of investigation, it is outside the scope of this program.
World Health Organization Classification
The World Health Organization (WHO) classification was revised in 2016 to simplify classification and define the role of molecular genetic testing (Table 1).1 An important change in MDS classification in the 2016 WHO classification was the creation of a category of MDS with ring sideroblasts (MDS-RS) with an SF3B1 mutation.16 If this mutation is present, a diagnosis of MDS-RS can be made with as few as 5% bone marrow ring sideroblasts; in the absence of an SF3B1 mutation, at least 15% bone marrow ring sideroblasts are required. Another noteworthy change was expansion of MDS with isolated del(5q) to include cases with one additional cytogenetic abnormality, as long as this second abnormality does not involve chromosome 7.16
| Table 1. World Health Organization Classification of Myelodysplastic Syndromes1 |
| Syndromes |
Dysplastic Lineages |
Cytopeniasa |
Ring Sideroblasts (% of marrow erythroid elements) |
Bone Marrow and Peripheral Blood Blasts |
Cytogenetics by Conventional Karyotype Analysis |
| MDS with single lineage dysplasia (MDS-SLD) |
1 |
1 or 2 |
<15% (<5%)b |
BM <5%, PB <1%, no Auer rods |
Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS with multilineage dysplasia (MDS-MLD) |
2 or 3 |
1–3 |
<15% (<5%)b |
BM <5%, PB <1%, no Auer rods |
Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS with ring sideroblasts (MDS-RS) |
| MDS-RS with single lineage dysplasia |
1 |
1 or 2 |
>15% (>5%)b |
BM <5%, PB <1%, no Auer rods |
Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS-RS with multilineage dysplasia |
2 or 3 |
1–3 |
>15% (>5%)b |
BM <5%, PB <1%, no Auer rods |
Any, unless fulfills all criteria for MDS with isolated del(5q) |
| Myelodysplastic syndrome associated with isolated del(5q) |
1–3 |
1–2 |
None or any |
BM <5%, PB <1%, no Auer rods |
del(5q) alone or with 1 additional abnormality except –7 or del(7q) |
| MDS with excess blasts (MDS-EB) |
| MDS-EB-1 |
0–3 |
1–3 |
None or any |
BM 5–9% or PB 2%–4%, no Auer rods |
Any |
| MDS-EB-2 |
0–3 |
1–3 |
None or any |
BM 10%–19% or PB 5%–19% or Auer rods |
Any |
| Myelodysplastic syndrome, unclassified |
| with 1% blood blasts |
1–3 |
1–3 |
None or any |
BM <5%, PB = 1%,c no Auer rods |
Any |
| with single lineage dysplasia and pancytopenia |
1 |
3 |
None or any |
BM <5%, PB <1%, no Auer rods |
Any |
| based on defining cytogenetic abnormality |
0 |
1–3 |
<15%d |
BM <5%, PB <1%, no Auer rods |
MDS-defining abnormality |
| Refractory cytopenia of childhood |
1–3 |
1–3 |
None |
BM <5%, PB <2% |
Any |
Abbreviations: BM, bone marrow; MDS, myelodysplastic syndromes; MDS-EB, myelodysplastic syndromes with excess blasts; MDS-RS, myelodysplastic syndromes with ring sideroblasts; PB, peripheral blood.
a Cytopenia defined as: hemoglobin <10 g/dL; platelet count <100 x 109/L; and absolute neutrophil count <1.8 x 109/L. Rarely, MDS may present with mild anemia or thrombocytopenia above these levels. Peripheral blood monocytes must be <1 x 109/L.
bSF3B1 mutation is present.
c 1% peripheral blood blasts must be recorded on at least 2 separate occasions.
d Cases with >15% ring sideroblasts by definition have significant erythroid dysplasia and are classified as MDS-RS-SLD. |
Prognostic Stratification
Scoring Systems
The International Prognostic Scoring System-Revised (IPSS-R) is based on analysis of more than 7,000 patients whose disease had not been treated with disease-altering therapy. It delineates patients into 5 prognostic categories that estimate OS ranging from 10 months to more than 8 years (Table 2).17 This model remains the standard risk assessment tool to guide initial therapy decisions in patients with MDS, though it may obscure the diversity in lower-risk MDS due to the propensity for clonal evolution.2,10
Machine learning was applied to MDS to develop a personalized risk-prediction model by synthesizing IPSS-R cytogenetic risk categories, 2008 WHO diagnosis, secondary versus de novo MDS, age, platelet count, white blood cell count, absolute lymphocyte, neutrophil and monocyte counts, hemoglobin level, percent bone marrow blasts, and mutational number and status for TP53, RUNX1, STAG2, ASXL1, SF3B1, SRSF2, RAD21, NRAS, NPM1, TET2, and EZH2.18 A web-based instrument is being developed that will predict probabilities for leukemic transformation and overall survival at different time points for an individual patient.
| Table 2. International Prognostic Scoring System—Revised for Myelodysplastic Syndromes17 |
| |
Points Assigned |
| Prognostic Variables |
0 |
0.5 |
1 |
1.5 |
2 |
3 |
4 |
| Cytogeneticsa |
Very good |
|
Good |
|
Intermediate |
Poor |
Very poor |
| Bone marrow blast (%) |
£2 |
|
>2 to <5 |
|
5–10 |
>10 |
|
| Hemoglobin (g/dL) |
³10 |
|
8 to <10 |
<8 |
|
|
|
| Platelets (cells/mm3) |
³100,000 |
50,000 to <100,000 |
<50,000 |
|
|
|
|
| Absolute neutrophil count (cells/mm3) |
³800 |
<800 |
|
|
|
|
|
aKaryotypes:
Very good (0 points): Y, del(11q)
Good (1 point): normal, del(5q), del(12p), del(20q), double including del(5q)
Intermediate (2 points): del(7q), +8, +19, i(17q), any other single or double independent clones
Poor (3 points): 7, inv(3)/t(3q)/del(3q), double, including 7/del(7q); complex: 3 abnormalities
Very poor (4 points): Complex: >3 abnormalities. |
| Score |
Risk Group |
Median Survival (years) |
| <1.5 |
Very low |
8.8 |
| >1.5–3 |
Low |
5.3 |
| >3–4.5 |
Intermediate |
3 |
| >4.5–6 |
High |
1.6 |
| >6 |
Very high |
0.8 |
Lower-Risk Versus Higher-Risk MDS
Treatment of MDS is generally directed at 2 broad groups based on prognosis determined by IPSS-R.9,10 Patients with very low, low, and intermediate risk with an IPSS-R score up to 3.5 are considered to have lower-risk MDS. Patients with IPSS-R score greater than 3.5 are managed as having higher-risk MDS.
GENERAL TREATMENT APPROACH IN LOWER-RISK MDS
The goals of therapy for patients with MDS depend on prognostic assessment and the individual patient profile including comorbid conditions and patient preferences. Universally, the goal of MDS therapy is delay of disease progression. In higher-risk patients, this may also include curative intent; however, management of lower-risk patients focuses more on symptom control, maintaining quality of life, and improvement of hematopoiesis.2,8 The only curative therapy for MDS is allogeneic stem cell transplant (SCT). Given the median age at diagnosis of MDS, SCT is used in fewer than 10% of patients.7
Supportive Care
The National Comprehensive Cancer Network (NCCN) Guidelines panel recommends supportive care, including clinical monitoring, psychosocial support, and quality-of-life assessment for all patients with MDS.10 Patients should receive antibiotics as treatment for infection. Routine antimicrobial prophylaxis and granulocyte colony stimulating factor (G-CSF) are not indicated in the absence of recurrent infections. HMAs or antithymocyte globulin (equine) (ATG) combined with cyclosporine warrant consideration for antimicrobial prophylaxis.2 Patients should receive platelet transfusions for thrombocytopenic bleeding and when platelet count is less than 10 x 109/L. Aminocaproic acid or other antifibrinolytic therapy may help control bleeding in patients with profound thrombocytopenia or those refractory to platelet transfusions.10
Timing of Treatment Initiation
Compelling data support delay of allogenic SCT in IPSS lower-risk patients. Markov model decision analyses demonstrated both younger patients receiving myeloablative conditioning and older patients receiving reduced-intensity conditioning have a reduced life expectancy with allogeneic SCT compared with conventional care.19,20
The benefit of early initiation of nontransplant strategies in lower-risk patients with MDS is unclear.21 Only 40% of patients with MDS in the United States receive drug therapy to manage these disorders.22 A systematic review of 1,314 patients did not discern an association with earlier treatment initiation and response in patients receiving erythropoiesis-stimulating agents (ESAs).23 Similarly, claims-based analysis of lenalidomide use in patients with MDS who were not yet transfusion dependent showed adverse events without an OS benefit from treatment before onset of transfusion need.24
Treatment algorithms for lower-risk MDS generally recommend initiation of therapy when patients have increased blasts or symptomatic cytopenias associated with MDS.10 The benefit of initiating HMAs or luspatercept before development of symptomatic anemia is unknown.
Management of Anemia
Red Blood Cell Transfusion
Patients with symptomatic anemia should receive red blood cell (RBC) transfusions.10 The use of transfusions should be minimized in patients without cardiovascular disease and those expected to be heavily transfused.10 Patients with lower-risk MDS have a relatively low risk for progression to acute myeloid leukemia and OS of 3 to 8 years17; therefore, they may survive long enough to develop complications associated with RBC transfusions. Even patients receiving only 1 RBC transfusion per month will receive more than 30 RBC transfusions over 3 years. Managing patients with RBC transfusions as the mainstay of therapy is associated with low median hemoglobin, poor quality of life, and possible iron overload.9
Erythropoiesis-Stimulating Agents
Current guidelines recommend use of ESAs to reduce the need for RBC transfusions in patients with lower-risk MDS.10 Unlike in some solid tumors,25 ESAs have not been shown to decrease OS or increase progression to leukemia in patients with MDS.10 Use of biosimilar epoetin alfa is acceptable to manage patients with MDS.26
Approximately 45% to 73% of lower-risk MDS patients respond to ESAs as initial therapy, with similar responses observed with epoetin and darbepoetin.27 Patients with lower-risk MDS who have a low percentage of bone marrow blasts, fewer than 2 somatic mutations, a serum erythropoietin (EPO) level less than 500 mU/mL (500 IU/L) and a history of receiving fewer than 2 units of RBC transfusions per month have the best chance at responding to ESAs.9,10
Compared with renal causes of anemia, higher doses of epoetin (30,000 to 60,000 units subcutaneously 1 to 2 times per week) and darbepoetin (100 to 300 mcg subcutaneously every other week) are required to achieve a response.9,10,28 Doses should be titrated up or down, to target a hemoglobin level of 10 to 12 g/dL. Responses may be delayed; patients should receive 8 to 12 weeks of treatment before discontinuing therapy because of lack of response.9
In patients with MDS, the duration of response to ESAs is typically 1 to 2 years.27 ESAs should be discontinued if there is no benefit or the response wanes.
Lenalidomide in MDS Patients With del(5q) Abnormality
Lenalidomide exerts direct effects on MDS cells by recruiting notably haploinsufficient substrates due to deletion 5q for ubiquitination.29,30 Lenalidomide is approved by the U.S. Food and Drug Administration (FDA) for treatment of transfusion-dependent anemia in patients with lower-risk MDS with isolated del(5q) or additional cytogenetic abnormalities. Red blood cell-transfusion independence (RBC-TI) for a minimum of 26 weeks was significantly improved in patients receiving lenalidomide 5 mg and 10 mg, 43% and 56%, respectively, versus placebo (6%) in a phase 3 randomized trial of patients with lower-risk MDS.31 Cytogenetic remission was achieved in 25% and 50% of patients receiving lenalidomide 5 mg and 10 mg, respectively, and subset analysis found patients achieving cytogenetic response or RBC-TI for more than 6 months correlated with improvement in OS.32
Two clinical trials have shown approximately one-fourth of patients with lower-risk MDS without a del(5q) abnormality achieve RBC-TI with lenalidomide.33,34 Patients in both of these trials were treatment naïve; however, the NCCN Guidelines panel recommends lenalidomide be considered for patients with symptomatic anemic associated with non-del(5q) MDS that does not respond to initial therapy.10
Immunosuppressive Therapy
The NCCN Guidelines panel recommends immunosuppressive therapy (IST) with ATG and cyclosporine for patients with MDS with a serum EPO greater than 500 mU/mL and a high likelihood of response to IST.10 Factors predictive of response to IST include age less than 60 years and fewer than 5% bone marrow blasts, patients with a hypocellular marrow and paroxysmal nocturnal hemoglobinuria clone positivity, and those with STAT-3 mutant cytotoxic T-cell clones.10
A phase 3 randomized controlled trial compared ATG and cyclosporine versus best supportive care patients with all IPSS risk categories of MDS.35 At 6 months, 29% of patients receiving IST achieved a hematologic response compared with 9% of patients receiving best supportive care; patients responding to IST had a median duration of response of 16.4 months. Despite the prolonged duration of response, no improvement in OS or time to progression to acute myeloid leukemia has been observed.2
Hypomethylating Agents
The benefit of HMAs in patients with lower-risk MDS is less clear than for patients with higher-risk MDS. The NCCN guidelines panel recommends initial therapy with azacitidine or decitabine in patients with a serum EPO level greater than 500 mU/mL who are unlikely to respond to IST.10
Azacitidine, administered subcutaneously, was evaluated in a phase 3, multicenter, randomized trial of patients diagnosed with any classification of MDS; 54% of patients had lower-risk MDS.36 Of the patients previously transfusion dependent who received azacitidine, 45% attained RBC-TI with a median time to treatment failure of 9.1 months.
Decitabine was evaluated in a multicenter, randomized phase 3 trial of patients diagnosed with MDS; approximately one-third of patients in this study had lower-risk MDS.37 Transfusion independence was seen in more than half of patients receiving decitabine for at least 6 cycles. A subsequent study evaluating outpatient intravenous decitabine administration once daily for 5 consecutive days in patients with any IPSS risk MDS reported RBC-TI in 33% of patients. Overall improvement was observed in 26% in patients with intermediate-1 IPSS risk, and the median duration of improvement was 10 months in all patients with a response.38
Decitabine combined with an oral cytidine deaminase inhibitor, cedazuridine, in a fixed dosing ratio reduces decitabine metabolism by intestinal cytidine deaminase and enables oral decitabine administration.39 On July 7, 2020, FDA approved the combination use of oral decitabine and cedazuridine for adult patients with intermediate-1, intermediate-2, and high-risk MDS based on 2 open-label, randomized, crossover trials and a phase 3 clinical trial, ASCERTAIN.40 The studies found similar drug concentrations in the 2 routes of administration of decitabine. A complete response was observed in 21% of patients receiving oral decitabine and cedazuridine and the median duration of response was 7.5 months.40,41
Oral azacitidine was evaluated in a phase 3 trial of patients with lower-risk MDS; however, the trial reported a higher incidence of early fatalities in patients receiving oral azacitidine, mostly from infection.42 Treatment of patients with MDS with oral azacitidine is not recommended outside clinical trials.43
Barriers to Anemia Management
Transfusion-Associated Morbidities
Risks associated with RBC transfusion include iron overload, transfusion-related acute lung injury (TRALI), and viral or bacterial infections.44,45 The use of blood-screening methods have decreased the infectious complications to almost negligible levels.45
While TRALI is a serious adverse event, its incidence is estimated at 1 in 5,000 transfusions. Both patients and providers express concern over the risks associated with excess iron resulting from multiple RBC transfusions.44 Excess iron from multiple RBC transfusions deposits in vital organs and may lead to cardiomyopathy, cirrhosis, diabetes mellitus, hypogonadism, and joint disease.46 Excess iron may exacerbate ineffective hematopoiesis in patients with MDS and is correlated with decreased OS in registry studies.47,48
The NCCN Guidelines panel recommends considering daily chelation with deferoxamine or deferasirox to decrease iron overload, particularly for patients who have lower-risk MDS or who are potential transplant candidates if more than 20 to 30 RBC transfusions have been received.10 Slow enrollment in the largest multicenter, randomized, placebo-controlled trial (TELESTO) of iron chelation in patients with lower-risk MDS led to modification of the study endpoints. The study was able to demonstrate chelation with deferasirox was associated with improvement in median event-free survival (EFS) by approximately 1 year over placebo.49
Refractory Response to ESA
ESAs are the most frequently employed treatment strategy in patients with lower-risk MDS with 73% of patients who receive therapy receiving an ESA.22 After 1 to 2 years of treatment, patients stop responding to ESAs.27 This leads to increased frequency of RBC transfusions, iron overload, decreased quality of life, and survival.10 Historically, fewer than 15% of patients who received initial therapy with an ESA received subsequent therapy after ESA discontinuation.22 Treatment strategies that are well tolerated by older patients with comorbidities and effective at decreasing transfusions are needed for patients with lower-risk MDS.
ESA-Refractory Anemia Management
Addition of Granulocyte Colony Stimulating Factor to ESAs
The addition of low-dose granulocyte colony stimulating factor (G-CSF) to ESA therapy has shown improvement in response to ESAs in several clinical trials.50-53 The NCCN Guidelines panel endorses addition of G-CSF in patients who have received 3 months of ESAs and have not responded or responded initially followed by loss of response.10 Predictive factors for response to combination therapy with epoetin and G-CSF include having a low serum EPO level (less than 200 mU/mL) at baseline, receiving 2 or fewer RBC transfusions/month, bone marrow blasts less than 10%, and MDS-RS.50-53
Luspatercept
While EPO stimulates RBC production, transforming growth factor b (TGF-b) inhibits RBC production.54 Luspatercept is a recombinant fusion protein that binds to endogenous TGF- b ligands to reduce Smad2/3 signaling and increase differentiation and proliferation of erythroid precursors.55
Luspatercept is recommended in the NCCN Guidelines for patients with symptomatic anemia who do not have a del(5q), present with ring sideroblasts greater than or equal to 15% or ring sideroblasts greater than or equal to 5% with an SF3B1 mutation, and have serum EPO levels of more than 500 mU/mL.10 Luspatercept is also recommended for similar patients with a serum EPO less than or equal to 500 mU/mL who do not respond to an ESAs combined with G-CSF.
These recommendations are based on a phase 3 trial in transfusion-dependent patients with lower-risk MDS with ring sideroblasts greater than or equal to 15%, or greater than or equal to 5% if an SF3B1 mutation was present, who were randomized to luspatercept or placebo administered every 3 weeks.56 Most patients (95%) had previously received ESAs. The primary endpoint of RBC-TI for 8 weeks or longer was achieved in 38% of patients receiving luspatercept and 13% of patients receiving placebo. Among patients who achieved RBC-TI, a response duration of 30.6 weeks was observed in patients receiving luspatercept.
Lenalidomide
Lenalidomide is recommended in the NCCN Guidelines for patients with symptomatic anemia who do not have a del(5q), have a serum EPO level less than or equal to 500 mU/mL, and did not respond to or progressed after therapy with an ESA.10 A phase 3 trial comparing lenalidomide to placebo in patients with lower-risk MDS who were refractory to or ineligible for ESA therapy, demonstrated that 27% of patients responded to ESA therapy compared with 2.5% of placebo patients.57 A response duration of 30.9 weeks was observed in patients who responded to lenalidomide.
A similar phase 3 trial randomized patients with lower-risk, transfusion-dependent MDS without a del(5q) with no response or relapse after an initial response to ESA therapy to receive lenalidomide plus epoetin vs lenalidomide monotherapy.58 Transfusion independence was achieved in 18.3% of patients receiving lenalidomide and 32% of patients receiving lenalidomide plus epoetin. Median response duration was 18.1 months for patients receiving lenalidomide and 15.1 months in patients receiving lenalidomide plus epoetin.
Hypomethylating Agents
A phase 2 study evaluated azacitidine in patients with lower-risk, transfusion-dependent MDS, with no response or relapse after an initial response to an ESA or ESA combined with G-CSF therapy.13 Of the 30 patients receiving at least 1 cycle of azacitidine monotherapy, 17% of patients achieved RBC-TI; 1 patient (6%) responded to a combination of azacitidine plus epoetin. The duration of response was less than 6 months in 4 of the 6 responding patients.
Another phase 2 study included patients with lower-risk, transfusion-dependent MDS who had relapsed after or were resistant to ESAs; it reported RBC-TI in 16.3% of patients receiving azacitidine and 14.3% of patients receiving azacitidine plus epoetin. The median duration of response in patients achieving RBC-TI was 7.6 months in patients receiving azacitidine and 9.7 months in those receiving azacitidine plus epoetin.59
A phase 2 study of azacitidine monotherapy in similar patients reported RBC RBC-TI in 33% of patients with a median duration of response of 10 months.60
FUTURE DIRECTIONS
Several promising agents with novel mechanisms of action are in clinical trials for patients with lower-risk MDS.54
Future studies of luspatercept include a phase 3 study randomizing patients with lower-risk MDS who require transfusion and have not received ESAs to luspatercept or epoetin (ClinicalTrials.gov Identifier: NCT03682536); completion of the primary endpoint is planned in 2021. A phase 1b/2 study combining luspatercept and lenalidomide in patients with lower-risk, non-del(5q) MDS for whom ESAs were ineffective or with a low likelihood of response was recently initiated (ClinicalTrials.gov Identifier: NCT04539236).
Roxadustat
Roxadustat inhibits hypoxia-inducible factor (HIF) prolyl hydroxylase, a major driver of erythropoiesis in the setting of hypoxia.54 A phase 3, randomized study comparing roxadustat to placebo in 184 patients with lower-risk MDS with a low RBC transfusion burden is expected to be completed in 2021 (ClinicalTrials.gov Identifier: NCT03263091). Preliminary results in 24 patients show RBC-TI for more than 8 weeks in 38% of patients.61 Results of a phase 2/3 study comparing roxadustat to placebo in 175 Chinese participants with lower-risk MDS are expected in late 2020 (ClinicalTrials.gov Identifier: NCT03303066).
Imetelstat
Imetelstat is a telomerase inhibitor that targets ineffective hematopoiesis in patients with MDS.54 A phase 2/3 study (IMerge) has been initiated to compare imetelstat to placebo in 225 patients with transfusion-dependent, lower-risk MDS that is refractory to ESA treatment (ClinicalTrials.gov Identifier: NCT 02598661). Preliminary results in 38 patients showed RBC-TI for more than 8 weeks in 42% of patients with a median duration of response of 20 months.62
Patient Case
A 61-year-old white man presented with increasing exercise intolerance and shortness of breath while playing tennis over the past 2 years. During evaluation by a cardiologist, he was noted to have anemia and he was referred to hematology. His hemoglobin was 7.9 g/dL, white blood cell count of 3.3 x 109/L and platelet count of 245 x 109/L. Bone marrow biopsy was consistent with myelodysplastic syndrome with ring sideroblasts (70%), multilineage dysplasia, and 1% blasts. He had normal cytogenetics. Next-generation sequencing detected mutations in SF3BI, ASXL1, DNMT3A and TET2. Splenomegaly was noted on computed topography scan of abdomen and pelvis. His EPO level was 571 mU/L.
The patient received luspatercept 1 mg/kg subcutaneously every 3 weeks for 2 doses. He remained transfusion dependent, and the dose was escalated to 1.33 mg/kg every 3 weeks. He achieved RBC-TI after 3 weeks at the new dose and continued every 3 weeks. He remained transfusion independent for 6 months before his hemoglobin declined, and he began to require 2 units of RBC transfusions every 2 weeks. At that time, the patient’s ferritin level was 1676 mcg/L, and he was initiated on deferasirox.
The patient was managed supportively with RBC transfusions for 6 months until he began to require 2 units of RBC every week. He was then started on azacitidine, and his transfusion needs decreased to 2 units of RBC every month. Bone marrow biopsy at that time was similar to previous results, with persistently elevated ring sideroblasts and no increase in blasts.
After 10 months of azacitidine, the patient’s transfusion needs again increased, requiring 2 units of RBCs weekly. Three years after his diagnosis, the patient had received more than 100 units of RBCs. His ferritin level was 2180 mcg/L, and liver biopsy revealed marked hepatocellular and Kupffer cell siderosis, mild pericellular fibrosis, and portal-based fibrosis. He developed nephrotoxicity from deferasirox and was diagnosed with stage 3 chronic kidney disease. Given his ongoing transfusion needs and relatively young age, the patient was referred for consideration of a reduced-intensity allogeneic SCT.
|
CONSIDERATIONS FOR PHARMACISTS
Patient Selection/Stratification
Approximately half of patients with MDS present with 1 or more comorbidities; assessment of performance status and comorbidities is helpful to gauge the ability of patients to tolerate therapy. Assessment of comorbidities can be done with various tools including the Charlson Comorbidity Index,63 Adult Comorbidity Evalution-27 (ACE-27),64 or the Hematopoietic Stem Cell Transplantation-Specific Comorbidity Index (HCT-CI).65 The NCCN Guidelines panel does not endorse a specific tool for comorbidities. The HCT-CI was predictive of OS and EFS in patients with lower-risk MDS; however, the ACE-27 was not.66,67
Once patients are determined to be suitable candidates for pharmacotherapy, treatment decisions for symptomatic anemia are guided by presence of a chromosome 5 deletion, ring sideroblasts with or without an SF3B1 mutation, and serum EPO level (Figure 1).2,10,68 Patients with a chromosome 5 deletion should receive lenalidomide. In general, patients with lower-risk MDS with a serum EPO less than 500 mU/L should receive initial therapy with an ESA. Luspatercept is appropriate for patients with at least 15% ring sideroblasts or with at least 5% ring sideroblasts in the presence of an SF3B1 mutation if they have a serum EPO level greater than 500 mU/L. Patients with a serum EPO level greater than 500 mU/L who are likely to respond to immunosuppression may benefit from ATG combined with cyclosporine. The remaining patients with lower-risk MDS may be candidates for azacitidine, decitabine, decitabine/cedazuridine, or lenalidomide.
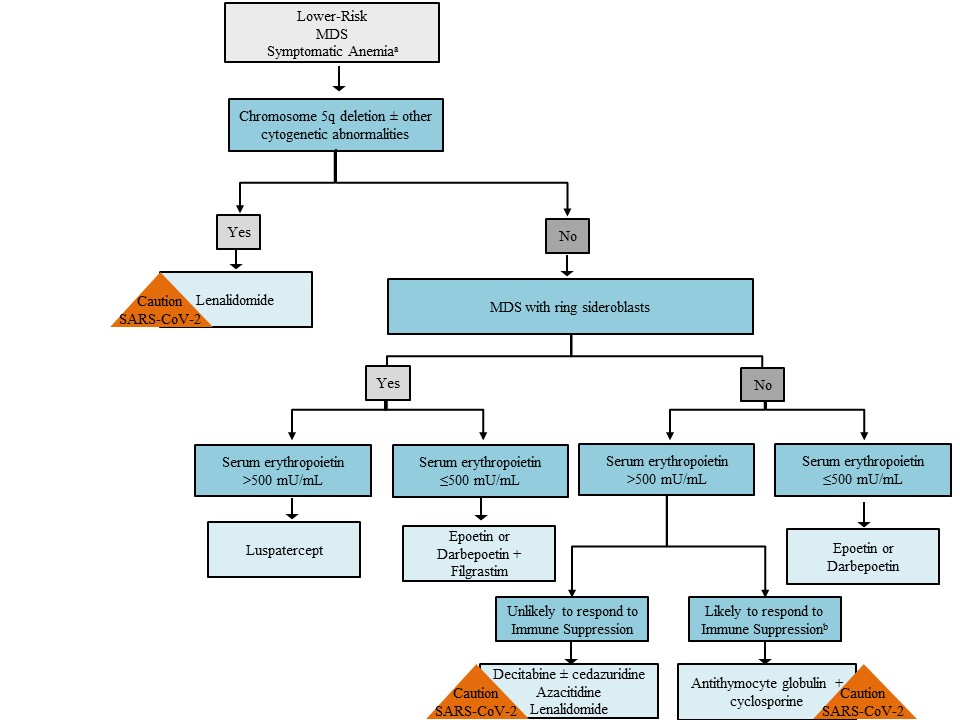
Dosing/Administration Considerations
Dosing guidelines for medications to manage anemia in lower-risk MDS are included in Table 3. In general, patients continue to receive medication as long as they maintain a benefit from therapy. Dose reduction for organ dysfunction is not needed for ESAs or luspatercept. Patients with renal dysfunction can receive lenalidomide and HMAs; however, treatment may be associated with a higher incidence of myelosuppression.69
Drug interactions are of limited concern with ESAs, HMAs, and lenalidomide. The newly approved combination of decitabine/cedazuridine provides an interesting theoretical concern about medications that are also metabolized by cytidine deaminase. Concomitant use of these medications in the presence of a cytidine deaminase inhibitor may increase their concentration.40 Examples of cytidine deaminase inhibitors would include: gemcitabine, capecitabine, cytarabine, zidovudine, telbivudine, didanosine, stavudine, lamivudine, abacavir, emtricitabine, entecavir, and tenofovir.
The coronavirus disease 2019 (COVID-19) pandemic, caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has accelerated home health options for a variety of parenteral medications for patients with cancer.70 ESAs may be administered in a health care facility or by the patient themselves, and the availability of premeasured syringes makes this an attractive option.71,72 Luspatercept requires reconstitution before administration; therefore, it is administered in a health care facility for the majority of patients, but home infusion services can be contracted for preparation and administration by a health care professional.55
Counseling Points
Pharmacists should review with patients instructions for taking decitabine/cedazuridine 35 mg/100 mg combination tablet at about the same time of day on the designated days of the cycle, and on an empty stomach, fasting for 2 hours before and after a dose.40 Tablets should not be chewed, crushed, or broken. Patients should be made aware of the teratogenic potential of lenalidomide and the requirements of the Risk Evaluation and Mitigation Strategy (REMS) program for this product.73
Reviews indicate that medication adherence and persistence for oral oncolytic therapy in patients with hematologic malignancies range from 50% to nearly 100%.74 Pharmacists should assess barriers to taking medication as prescribed and continuing therapy over time by emphasizing the importance of therapy in management of lower-risk MDS.
Copayment assistance programs are available for luspatercept, lenalidomide, and decitabine/cedazuridine.75,76 Patients and providers should review individual company websites for eligibility criteria; restrictions apply for most programs.
Monitoring/Adverse Effect Management
All patients should have routine monitoring with complete blood counts to determine both response and toxicity. Patients receiving azacitidine and lenalidomide should have renal function monitored as well.
Patients receiving lenalidomide, azacitidine, and decitabine may develop neutropenia and/or thrombocytopenia when initiating therapy.9 Treatment-induced neutropenia may be managed by holding treatment, dose reductions, or use of G-CSF. Treatment-induced thrombocytopenia is similarly managed by holding treatment, dose reductions, or platelet transfusion.
Patients receiving ESAs or lenalidomide may develop thromboembolic complications; however, prophylaxis with aspirin or anticoagulants is generally not indicated in patients with MDS. Antiemetic prophylaxis is recommended for both azacitidine and decitabine. Azacitidine-induced erythema at the site of subcutaneous injection may be minimized with the use of hot or cold compresses or topical corticosteroids.
| Table 3. Dosing, Administration and Safety of First-Line Agents to Manage Anemia in Myelodysplastic Syndromes40,71-73,77,78 |
| Drug and Regimen |
Adjustment in Organ Dysfunction |
Warnings and Adverse Effects |
Azacitidine
75 mg/m2 IV or SQ once daily for 7 days every 4 weeks; dose may be increased to 100 mg/m2 if no beneficial effect after 2 cycles and no toxicity
Azacitidine 75 mg/m2 IV or SQ daily for 5 days, followed by 2 days no treatment, then 75 mg/m2 for 2 days79 |
Hepatic impairment: no adjustment
CrCL ≥30 mL/min: no dose adjustment is needed
CrCL <30 mL/min: no dose adjustment is needed
for initial cycle, metabolites are renally excreted and adjustment may be needed69,80,81 |
Warnings and precautions: anemia, neutropenia, thrombocytopenia
Contraindications: advanced malignant hepatic tumors
Adverse effects: nausea, anemia, thrombocytopenia, vomiting, pyrexia, leukopenia, diarrhea, injection site erythema, constipation, neutropenia, ecchymosis
REMS: none |
Darbepoetin
100 to 300 mcg SQ every other week |
Hepatic impairment: no adjustment
Renal impairment: no adjustment |
Boxed warning: increased mortality, tumor progression or recurrence in some tumor types, serious cardiovascular events, thromboembolic events, stroke
Contraindications: uncontrolled hypertension; hypersensitivity to product
Adverse effects: edema, dizziness, headache, diarrhea, arthralgia, rash, hypertension, thrombotic events
REMS: FDA in 2017 determined REMS program no longer required |
Decitabine
20 mg/m2 IV daily for 5 days, every 4 weeks38 |
Hepatic impairment: no adjustment before initiation of therapy. Hold therapy for ALT and/or bilirubin ≥2 times ULN
Renal impairment: no adjustment before initiation of therapy. Hold therapy for serum creatinine ≥2 mg/dL |
Warnings and precautions: neutropenia, thrombocytopenia, embryo-fetal toxicity
Contraindications: none
Adverse effects: anemia, febrile neutropenia, thrombocytopenia, neutropenia, nausea, diarrhea, constipation, fatigue, edema, pyrexia, pneumonia |
| Decitabine 35 mg/cedazuridine 100 mg 1 tablet orally on days 1 through 5, every 4 weeks |
Mild hepatic impairment (total bilirubin >1.5 times ULN or AST > ULN): no adjustment
Hold therapy for ALT and/or bilirubin >2 times ULN
CrCL >30 mL/min: no adjustment before initiation of therapy. Hold therapy for serum creatinine ≥2 mg/dL |
Adverse effects: fatigue, constipation, hemorrhage, myalgia, mucositis, arthralgia, nausea, dyspnea, diarrhea, rash, dizziness, febrile neutropenia, edema, headache, cough, decreased appetite, upper respiratory tract infection, pneumonia-elevated transaminases
REMS: none |
Epoetin alfa
30,000 to 60,000 units SQ 1 to 2 times per week |
Hepatic impairment: no adjustment
Renal impairment: no adjustment |
Boxed warning: increased mortality, tumor progression or recurrence in some tumor types, myocardial infarction, stroke, venous thromboembolism, thrombosis of vascular access
Contraindications: uncontrolled hypertension; pure red cell aplasia that begins after treatment with ESAs, hypersensitivity to product, use of the multidose vials containing benzyl alcohol in pregnant or lactating women
Adverse effects: nausea, myalgia, hypertension, thrombotic events
REMS: FDA in 2017 determined REMS program no longer required |
Lenalidomide
10 mg orally once daily |
Mild-to-severe hepatic impairment: no adjustment
CrCL 30–60 mL/min: 5 mg once daily
CrCL <30 mL/min (not requiring dialysis): 2.5 mg once daily
CrCL <30 mL/min (requiring dialysis): 2.5 mg once daily. On dialysis days administer dose after dialysis
Dose reduction for neutropenia and thrombocytopenia that develops while on therapy |
Boxed warning: embryo-fetal toxicity, hematologic toxicity, venous and arterial thromboembolism
Warnings and precautions: second primary malignancies, hepatotoxicity, severe cutaneous reactions, tumor lysis syndrome, tumor flare reaction, impaired stem cell mobilization, hypersensitivity, increased mortality in some tumor types
Adverse events: thrombocytopenia, neutropenia, diarrhea, pruritus, rash, fatigue, constipation, nausea, nasopharyngitis, arthralgia, pyrexia, back pain, edema, cough, dizziness, headache, muscle cramp, dyspnea, pharyngitis, epistaxis
REMS: REMS program required |
Luspatercept-aamt
1 mg/kg SQ every 3 weeks |
Mild-to-severe hepatic impairment: no adjustment
Mild-to-moderate renal impairment: no adjustment |
Warnings and precautions: thrombosis/thromboembolism; hypertension; embryo-fetal toxicity
Contraindications: none
Adverse effects: headache, bone pain, arthralgia, fatigue, cough, abdominal pain, diarrhea, dizziness
REMS: none |
| Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; CrCL, creatinine clearance, ESAs, erythropoiesis-stimulating agents; FDA, Food and Drug Administration; intravenous, IV; REMS, risk evaluation and management strategy; subcutaneous, SQ; ULN, upper limit of normal. |
COVID-19 PANDEMIC CONSIDERATIONS
Reported case fatality rates from SARS-CoV-2 have been as high as 37% in patients with hematologic malignancies.82 In general, the American Society of Clinical Oncology (ASCO) and the NCCN recommend patients receive critical anticancer therapy during the COVID-19 pandemic.83,84
An international expert panel for patients with acute leukemias and myeloid neoplasms recommends that most patients with lower-risk MDS be managed with a watch-and-wait approach during the COVID-19 pandemic.68 ESAs for ESA-naïve patients and luspatercept should be considered to minimize the need for RBC transfusions. The panel suggests deferring initiation of lenalidomide in newly diagnosed patients with del(5q) because of the risk for myelosuppression during therapy initiation (Figure 1).
Other considerations for altering cancer therapy during the pandemic relevant to patients with MDS include switching from intravenous to oral therapies (e.g., decitabine with cedazuridine instead of intravenous decitabine) with enhanced monitoring of patients in their homes to decrease clinic visits; consideration of alternative dosing schedules to allow for fewer in-person visits to the cancer center and/or the infusion center (e.g., dispensing 56-day supply of lenalidomide for patients already on therapy); and transitioning outpatient care to care at home whenever possible (e.g., administration of growth factors).83,84
The COVID-19 pandemic has made digital health care a more viable alternative with the launch of new remote service options including video visits, text, email, and mobile-phone applications.85
CONCLUSION
The treatment of MDS is rapidly changing, with the approval of 2 new agents in 2020 alone. As more is understood about the clonal evolution of MDS, pharmacists need to understand how to tailor treatment selection for patients with MDS based on risk factors outside of traditional cytopenias. Additionally, management will remain contingent on the complex nature of these patients due to advanced age, comorbid conditions, and inability to tolerate intensive therapies.10
If MDS progresses to acute myeloid leukemia, patients experience lower response rates to standard therapy than patients with de novo acute myeloid leukemia, creating a treatment conundrum.10 Advanced age, significant comorbidities, low hemoglobin levels, RBC transfusion needs, and female sex are associated with poor health-related quality of life in patients with MDS.24
Pharmacists’ knowledge about toxicities, monitoring parameters, and dose adjustments in the setting of adverse effects is paramount to ensuring excellent patient care. Improved management of anemia and reduced need for RBC transfusions contribute to improvement or maintenance of quality of life in patients with MDS.
Figure 1. General Approach to Lower-Risk Myelodysplastic Syndromes With Anemia2,10,68
All patients with lower-risk MDS dominated by anemia should receive supportive care with transfusions and antimicrobial therapy as needed. The initial therapeutic choice depends on the karyotype, presence of MDS with ring sideroblasts (MDS-RS) subtype, and the serum erythropoietin (EPO) level. For patients with del(5q), lenalidomide is an appropriate first choice. Luspatercept is appropriate initial therapy for patients with MDS-RS if they have a serum EPO level greater than 500 mU/L. For patients without del(5q) or MDS-RS, but with EPO levels less than 500 U/L, the erythropoiesis-stimulating agents (ESAs) epoetin and darbepoetin are recommended. Immunosuppressive therapy should be considered for patients likely to respond (i.e., patients ≤60 years with ≤5% marrow blasts; patients with a hypocellular marrow, PNH clone positivity, or STAT-mutant cytotoxic T-cell clone). Lenalidomide or hypomethylating agents (HMAs) are appropriate therapy for patients who do not fall into the populations described above. During the COVID-19 pandemic, experts recommend avoiding initiating therapy with lenalidomide or HMAs because of the risk for neutropenia and potential for increased viral exposure in the health care setting. Similarly, patients receiving immunosuppressive therapy may be at increased risk for complications from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
Abbreviations: MDS, myelodysplastic syndromes; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
a All patients should be offered best supportive care and can be considered for enrollment in a clinical trial at any time.
b Likely to respond to Immune Suppression includes patients ≤60 years with ≤5% marrow blasts; patients with a hypocellular marrow, PNH clone positivity, or STAT- mutant cytotoxic T-cell clone.
Updated: August 31, 2021
- Venetoclax in combination with azacitidine has been granted Breakthrough Therapy designation by the US Food and Drug Administration for the treatment of adult patients with previously untreated intermediate-, high-, and very high–risk myelodysplastic syndromes (MDS) based on the revised International Prognostic Scoring System. This designation was granted based on interim results from the M15-531 study. Based on these data, a phase III clinical trial (VERONA) was launched comparing azacitidine plus venetoclax with azacitidine plus placebo for the treatment of higher-risk MDS.
Reference:
Garcia JS, Wei AH, Borate U, et al: Safety, efficacy, and patient-reported outcomes of venetoclax in combination with azacitidine for the treatment of patients with higher-risk myelodysplastic syndrome: A phase 1b study. 2020 ASH Annual Meeting & Exposition. Abstract 656. Presented December 7, 2020.
REFERENCES
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi:10.1182/blood-2016-03-643544
- Garcia-Manero G, Chien K, Montalban-Bravo G. Myelodysplastic syndromes: 2021 update on diagnosis, risk-stratification and management. Am J Hematol. Published online August 3, 2020. doi:10.1002/ajh.25950
- Ma X. Epidemiology of myelodysplastic syndromes. Am J Med. 2012;125(7 suppl):S2–S5. doi:10.1016/j.amjmed.2012.04.014
- Zeidan AM, Shallis RM, Wang R, Davidoff A, Ma X. Epidemiology of myelodysplastic syndromes: why characterizing the beast is a prerequisite to taming it. Blood Rev. 2019;34:1–15. doi:10.1016/j.blre.2018.09.001
- Cogle CR. Incidence and burden of the myelodysplastic syndromes. Curr Hematol Malig Rep. 2015;10(3):272–281. doi:10.1007/s11899-015-0269-y
- Gangat N, Patnaik MM, Begna K, et al. Survival trends in primary myelodysplastic syndromes: a comparative analysis of 1000 patients by year of diagnosis and treatment. Blood Cancer J. 2016;6(4):e414. doi:10.1038/bcj.2016.23
- Steensma DP. Myelodysplastic syndromes: diagnosis and treatment. Mayo Clin Proc. 2015;90(7):969–983. doi:10.1016/j.mayocp.2015.04.001
- Platzbecker U. Treatment of MDS. Blood. 2019;133(10):1096–1107. doi:10.1182/blood-2018-10-844696
- Fenaux P, Platzbecker U, Ades L. How we manage adults with myelodysplastic syndrome. Br J Haematol. 2020;189(6):1016–1027. doi:10.1111/bjh.16206
- NCCN Clinical Practice Guidelnies in Oncology (NCCN Guidelines). Myelodysplastic Syndromes. Version 2.2021. Updated September 11, 2020. Accessed September 18, 2020. https://www.nccn.org/professionals/physician_gls/pdf/mds.pdf.
- Ogawa S. Genetics of MDS. Blood. 2019;133(10):1049–1059. doi:10.1182/blood-2018-10-844621
- Chokr N, Pine AB, Bewersdorf JP, Shallis RM, Stahl M, Zeidan AM. Getting personal with myelodysplastic syndromes: is now the right time? Expert Rev Hematol. 2019;12(4):215–224. doi:10.1080/17474086.2019.1592673
- Tobiasson M, Dybedahl I, Holm MS, et al. Limited clinical efficacy of azacitidine in transfusion-dependent, growth factor-resistant, low- and Int-1-risk MDS: results from the nordic NMDSG08A phase II trial. Blood Cancer J. 2014;4(3):e189. doi:10.1038/bcj.2014.8
- Bejar R, Levine R, Ebert BL. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J Clin Oncol. 2011;29(5):504–515. doi:10.1200/JCO.2010.31.1175
- Gondek LP, Dezern AE. Assessing clonal haematopoiesis: clinical burdens and benefits of diagnosing myelodysplastic syndrome precursor states. Lancet Haematol. 2020;7(1):e73–e81. doi:10.1016/s2352-3026(19)30211-x
- Hasserjian RP, Kelley TW, Weinberg OK, Morgan EA, Fend F. Genetic testing in the diagnosis and biology of myeloid neoplasms (excluding acute leukemias). Am J Clin Pathol. 2019;152(3):302–321. doi:10.1093/ajcp/aqz069
- Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454–2465. doi:10.1182/blood-2012-03-420489
- Nazha A, Komrokji RS, Meggendorfer M, et al. A Personalized prediction model to risk stratify patients with myelodysplastic syndromes. Blood. 2018;132(suppl 1):793. doi:10.1182/blood-2018-99-114774
- Cutler CS, Lee SJ, Greenberg P, et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood. 2004;104(2):579–585. doi:10.1182/blood-2004-01-0338
- Koreth J, Pidala J, Perez WS, et al. Role of reduced-intensity conditioning allogeneic hematopoietic stem-cell transplantation in older patients with de novo myelodysplastic syndromes: an international collaborative decision analysis. J Clin Oncol. 2013;31(21):2662–2670. doi:10.1200/JCO.2012.46.8652
- Steensma DP. Does early diagnosis and treatment of myelodysplastic syndromes make a difference? Best Pract Res Clin Haematol. 2019;32(4):101099. doi:10.1016/j.beha.2019.101099
- Ma X, Steensma DP, Scott BL, Kiselev P, Sugrue MM, Swern AS. Selection of patients with myelodysplastic syndromes from a large electronic medical records database and a study of the use of disease-modifying therapy in the United States. BMJ Open. 2018;8(7):e019955. doi:10.1136/bmjopen-2017-019955
- Moyo V, Lefebvre P, Duh MS, Yektashenas B, Mundle S. Erythropoiesis-stimulating agents in the treatment of anemia in myelodysplastic syndromes: a meta-analysis. Ann Hematol. 2008;87(7):527–536. doi:10.1007/s00277-008-0450-7
- Brunner AM, Weng S, Cronin A, et al. Impact of lenalidomide use among non-transfusion dependent patients with myelodysplastic syndromes. Am J Hematol. 2018;93(9):1119–1126. doi:10.1002/ajh.25166
- Bohlius J, Schmidlin K, Brillant C, et al. Recombinant human erythropoiesis-stimulating agents and mortality in patients with cancer: a meta-analysis of randomised trials. Lancet. 2009;373(9674):1532–1542. doi:10.1016/S0140-6736(09)60502-X
- Castelli R, Deliliers GL, Colombo R, Moreo G, Gallipoli P, Pantaleo G. Biosimilar epoetin in elderly patients with low-risk myelodysplastic syndromes improves anemia, quality of life, and brain function. Ann Hematol. 2014;93(9):1523–1529. doi:10.1007/s00277-014-2070-8
- Park S, Greenberg P, Yucel A, et al. Clinical effectiveness and safety of erythropoietin-stimulating agents for the treatment of low- and intermediate-1-risk myelodysplastic syndrome: a systematic literature review. Br J Haematol. 2019;184(2):134–160. doi:10.1111/bjh.15707
- Balleari E, Filiberti RA, Salvetti C, et al. Effects of different doses of erythropoietin in patients with myelodysplastic syndromes: a propensity score-matched analysis. Cancer Med. 2019;8(18):7567–7576. doi:10.1002/cam4.2638
- Stahl M, Zeidan AM. Lenalidomide use in myelodysplastic syndromes: insights into the biologic mechanisms and clinical applications. Cancer. 2017;123(10):1703–1713. doi:10.1002/cncr.30585
- Fink EC, Ebert BL. The novel mechanism of lenalidomide activity. Blood. 2015;126(21):2366–2369. doi:10.1182/blood-2015-07-567958
- Fenaux P, Giagounidis A, Selleslag D, et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with low-/intermediate-1-risk myelodysplastic syndromes with del5q. Blood. 2011;118(14):3765–3776. doi:10.1182/blood-2011-01-330126
- Sanchez-Garcia J, Del Canizo C, Lorenzo I, et al. Multivariate time-dependent comparison of the impact of lenalidomide in lower-risk myelodysplastic syndromes with chromosome 5q deletion. Br J Haematol. 2014;166(2):189–201. doi:10.1111/bjh.12876
- Garcia-Manero G, Almeida A, Fenaux P, et al. Clinical benefit-risk profile of lenalidomide in patients with lower-risk myelodysplastic syndromes without del(5q): results of a phase III trial. Clin Lymphoma Myeloma Leuk. 2019;19(4):213–219 e4. doi:10.1016/j.clml.2018.12.012
- Raza A, Reeves JA, Feldman EJ, et al. Phase 2 study of lenalidomide in transfusion-dependent, low-risk, and intermediate-1 risk myelodysplastic syndromes with karyotypes other than deletion 5q. Blood. 2008;111(1):86–93. doi:10.1182/blood-2007-01-068833
- Passweg JR, Giagounidis AA, Simcock M, et al. Immunosuppressive therapy for patients with myelodysplastic syndrome: a prospective randomized multicenter phase III trial comparing antithymocyte globulin plus cyclosporine with best supportive care—SAKK 33/99. J Clin Oncol. 2011;29(3):303–309. doi:10.1200/JCO.2010.31.2686
- Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20(10):2429–2440. doi:10.1200/JCO.2002.04.117
- Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106(8):1794–1803. doi:10.1002/cncr.21792
- Steensma DP, Baer MR, Slack JL, et al. Multicenter study of decitabine administered daily for 5 days every 4 weeks to adults with myelodysplastic syndromes: the alternative dosing for outpatient treatment (ADOPT) trial. J Clin Oncol. 2009;27(23):3842–3848. doi:10.1200/JCO.2008.19.6550
- Dacagen. Prescribing information. Otsuka Pharmaceutical Co., Ltd; 2020. Accessed August 2, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/021790s025lbl.pdf.
- Inqovi. Prescribing information. Otsuka Pharmaceutical Co., Ltd; 2020. Accessed September 28, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212576s000lbl.pdf.
- Garcia-Manero G, Griffiths EA, Steensma DP, et al. Oral cedazuridine/decitabine for MDS and CMML: a phase 2 pharmacokinetic/pharmacodynamic randomized crossover study. Blood. 2020;136(6):674–683. doi:10.1182/blood.2019004143
- Garcia-Manero G, Santini V, Almeida A, et al. A phase III placebo-controlled trial of CC-486 in patients with red blood cell transfusion-dependent anemia and thrombocytopenia due to IPSS lower-risk myelodysplastic syndromes. European Hematology Association Virtual Congress. Abstract S180. https://library.ehaweb.org/eha/2020/eha25th/295000/guillermo.garcia-manero.a.phase.iii.placebo-controlled.trial.of.cc-486.in.html?f=listing%3D0%2Abrowseby%3D8%2Asortby%3D2%2Asearch%3Dblast.
- Onureg. Prescribing information. Celgene Corporation; 2020. Accessed September 28, 2020. https://packageinserts.bms.com/pi/pi_onureg.pdf.
- King D, Schmidt R, Lesser M, Houk A, Salkeld E, Cogle CR. Patient and physician perceptions about blood transfusions in the myelodysplastic syndromes. Leuk Res. 2020;96:106425. doi:10.1016/j.leukres.2020.106425
- Sahu S, Hemlata, Verma A. Adverse events related to blood transfusion. Indian J Anaesth. 2014;58(5):543–551. doi:10.4103/0019-5049.144650
- Moukalled NM, El Rassi FA, Temraz SN, Taher AT. Iron overload in patients with myelodysplastic syndromes: an updated overview. Cancer. 2018;124(20):3979–3989. doi:10.1002/cncr.31550
- Hoeks M, Yu G, Langemeijer S, et al. Impact of treatment with iron chelation therapy in patients with lower-risk myelodysplastic syndromes participating in the European MDS registry. Haematologica. 2020;105(3):640–651. doi:10.3324/haematol.2018.212332
- Leitch HA, Parmar A, Wells RA, et al. Overall survival in lower IPSS risk MDS by receipt of iron chelation therapy, adjusting for patient-related factors and measuring from time of first red blood cell transfusion dependence: an MDS-CAN analysis. Br J Haematol. 2017;179(1):83–97. doi:10.1111/bjh.14825
- Angelucci E, Li J, Greenberg P, et al. Iron chelation in transfusion-dependent patients with low- to intermediate-1-risk myelodysplastic syndromes: a randomized trial. Ann Intern Med. 2020;172(8):513–522. doi:10.7326/M19-0916
- Jadersten M, Malcovati L, Dybedal I, et al. Erythropoietin and granulocyte-colony stimulating factor treatment associated with improved survival in myelodysplastic syndrome. J Clin Oncol. 2008;26(21):3607–3613. doi:10.1200/JCO.2007.15.4906
- Kelaidi C, Beyne-Rauzy O, Braun T, et al. High response rate and improved exercise capacity and quality of life with a new regimen of darbepoetin alfa with or without filgrastim in lower-risk myelodysplastic syndromes: a phase II study by the GFM. Ann Hematol. 2013;92(5):621–631. doi:10.1007/s00277-013-1686-4
- Park S, Grabar S, Kelaidi C, et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood. 2008;111(2):574–582. doi:10.1182/blood-2007-06-096370
- Greenberg PL, Sun Z, Miller KB, et al. Treatment of myelodysplastic syndrome patients with erythropoietin with or without granulocyte colony-stimulating factor: results of a prospective randomized phase 3 trial by the Eastern Cooperative Oncology Group (E1996). Blood. 2009;114(12):2393–2400. doi:10.1182/blood-2009-03-211797
- Bewersdorf JP, Zeidan AM. Evolving therapies for lower-risk myelodysplastic syndromes. Ann Hematol. 2020;99(4):677–692. doi:10.1007/s00277-020-03963-1
- Reblozyl. Prescribing information. Celgene Corporation; 2019. Accessed September 18, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761136lbl.pdf.
- Fenaux P, Platzbecker U, Mufti GJ, et al. Luspatercept in patients with lower-risk myelodysplastic syndromes. N Engl J Med. Jan 9 2020;382(2):140–151. doi:10.1056/NEJMoa1908892
- Santini V, Almeida A, Giagounidis A, et al. Randomized phase III study of lenalidomide versus placebo in RBC transfusion-dependent patients with lower-risk non-del(5q) myelodysplastic syndromes and ineligible for or refractory to erythropoiesis-stimulating agents. J Clin Oncol. 2016;34(25):2988–2996. doi:10.1200/JCO.2015.66.0118
- Toma A, Kosmider O, Chevret S, et al. Lenalidomide with or without erythropoietin in transfusion-dependent erythropoiesis-stimulating agent-refractory lower-risk MDS without 5q deletion. Leukemia. 2016;30(4):897–905. doi:10.1038/leu.2015.296
- Thepot S, Ben Abdelali R, Chevret S, et al. A randomized phase II trial of azacitidine ± epoetin-beta in lower-risk myelodysplastic syndromes resistant to erythropoietic stimulating agents. Haematologica. 2016;101(8):918–925. doi:10.3324/haematol.2015.140988
- Fili C, Malagola M, Follo MY, et al. Prospective phase II study on 5-days azacitidine for treatment of symptomatic and/or erythropoietin unresponsive patients with low/INT-1-risk myelodysplastic syndromes. Clin Cancer Res. 2013;19(12):3297–3308. doi:10.1158/1078-0432.CCR-12-3540
- Yu K-HP, Robert L, Bartels P, et al. Roxadustat (FG4592; ASP1517; AZD9941) in the treatment of anemia in patients with lower risk myelodysplastic syndrome (LR-MDS) and low red blood cell (RBC) transfusion burden (LTB). Blood. 2019;134(suppl 1):843. doi:10.1182/blood-2019-128714
- Platzbecker U, Fenaux P, Steensma DP, et al. Treatment with imetelstat provides durable transfusion independence in heavily transfused non-del(5q) lower risk MDS relapsed/refractory to erythropoiesis stimulating agents. European Hematology Association Virtual Congress. Abstract S183. https://library.ehaweb.org/eha/2020/eha25th/295003/uwe.platzbecker.treatment.with.imetelstat.provides.durable.transfusion.html?f=listing%3D0%2Abrowseby%3D8%2Asortby%3D1%2Asearch%3Ds183.
- Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373–383. doi:10.1016/0021-9681(87)90171-8
- Naqvi K, Garcia-Manero G, Sardesai S, et al. Association of comorbidities with overall survival in myelodysplastic syndrome: development of a prognostic model. J Clin Oncol. 2011;29(16):2240–2246. doi:10.1200/JCO.2010.31.3353
- Sorror ML, Storb RF, Sandmaier BM, et al. Comorbidity-age index: a clinical measure of biologic age before allogeneic hematopoietic cell transplantation. J Clin Oncol. 2014;32(29):3249–3256. doi:10.1200/JCO.2013.53.8157
- Sperr WR, Wimazal F, Kundi M, et al. Comorbidity as prognostic variable in MDS: comparative evaluation of the HCT-CI and CCI in a core dataset of 419 patients of the Austrian MDS Study Group. Ann Oncol. 2010;21(1):114–149. doi:10.1093/annonc/mdp258
- Daver N, Naqvi K, Jabbour E, et al. Impact of comorbidities by ACE-27 in the revised-IPSS for patients with myelodysplastic syndromes. Am J Hematol. 2014;89(5):509–516. doi:10.1002/ajh.23675
- Zeidan AM, Boddu PC, Patnaik MM, et al. Special considerations in the management of adult patients with acute leukaemias and myeloid neoplasms in the COVID-19 era: recommendations from a panel of international experts. Lancet Haematol. 2020;7(8):e601–e612. doi:10.1016/S2352-3026(20)30205-2
- Batty GN, Kantarjian H, Issa JP, et al. Feasibility of therapy with hypomethylating agents in patients with renal insufficiency. Clin Lymphoma Myeloma Leuk. 2010;10(3):205–210. doi:10.3816/CLML.2010.n.032
- Cavallo J. How the COVID-19 pandemic is propelling the delivery of home care for patients with cancer. Updated May 25, 2020. Accessed October 8, 2020. https://ascopost.com/issues/may-25-2020/how-the-covid-19-pandemic-is-propelling-the-delivery-of-home-care-for-patients-with-cancer/.
- Aranesp. Prescribing information. Amgen Inc.; 2019. Accessed September 28, 2020. https://www.pi.amgen.com/~/media/amgen/repositorysites/pi-amgen-com/aranesp/ckd/aranesp_pi_hcp_english.pdf.
- Epogen. Prescribing information. Amgen Inc.; 2018. Accessed September 27, 2020. https://www.pi.amgen.com/~/media/amgen/repositorysites/pi-amgen-com/epogen/epogen_pi_hcp_english.pdf.
- Revlimid. Prescribing information. Celgene Corporation; 2019. Accessed September 27, 2020. https://packageinserts.bms.com/pi/pi_revlimid.pdf.
- Rosenberg SM, Petrie KJ, Stanton AL, Ngo L, Finnerty E, Partridge AH. Interventions to enhance adherence to oral antineoplastic agents: a scoping review. J Natl Cancer Inst. 2020;112(5):443–465. doi:10.1093/jnci/djz244
- Celgene Patient Support. Celgene Corporation. Updated June 2020. Accessed October 8, 2020. https://celgenepatientsupport.com/hcp/find-financial-help/.
- Taiho Oncology Patient Support. Taiho Oncology, Inc. Updated July 2020. Accessed October 8, 2020. https://www.taihopatientsupport.com/.
- Vidaza. Prescribing information. Celgene Corporation; 2020. Accessed September 28, 2020. https://packageinserts.bms.com/pi/pi_vidaza.pdf.
- Dacogen. Prescribing information. Otsuka America Pharmaceutical, Inc; 2020. Accessed September 28, 2020. https://www.otsuka-us.com/media/static/DACOGEN-PI.pdf.
- Lyons RM, Cosgriff TM, Modi SS, et al. Hematologic response to three alternative dosing schedules of azacitidine in patients with myelodysplastic syndromes. J Clin Oncol. 2009;27(11):1850–1856. doi:10.1200/JCO.2008.17.1058
- Douvali E, Papoutselis M, Spanoudakis E, et al. Safety and efficacy of azacitidine in myelodysplastic syndrome (MDS) patients with mild and moderate renal impairment. Blood. 2012;120(21):1716–1716. doi:10.1182/blood.V120.21.1716.1716
- Laille E, Goel S, Mita AC, et al. A phase I study in patients with solid or hematologic malignancies of the dose proportionality of subcutaneous azacitidine and its pharmacokinetics in patients with severe renal impairment. Pharmacotherapy. 2014;34(5):440–451. doi:10.1002/phar.1371
- Mehta V, Goel S, Kabarriti R, et al. Case fatality rate of cancer patients with COVID-19 in a New York hospital system. Cancer Discov. 2020;10(7):935–941. doi:10.1158/2159-8290.CD-20-0516
- COVID-19 Patient Care Information. News release. American Society of Clinical Oncology. April 23, 2020. Accessed October 13, 2020. https://www.asco.org/asco-coronavirus-information/care-individuals-cancer-during-covid-19
- Cinar P, Kubal T, Freifeld A, et al. Safety at the time of the COVID-19 pandemic: how to keep our oncology patients and healthcare workers safe. J Natl Compr Canc Netw. April 15, 2020. doi:10.6004/jnccn.2020.7572
- Keesara S, Jonas A, Schulman K. Covid-19 and health care's digital revolution. N Engl J Med. 2020;382(23):e82. doi:10.1056/NEJMp2005835